
Abstract
The bacterial endotoxin test, which uses amebocyte lysate reagents of horseshoe crab origin, is a sensitive, reproducible and simple assay to measure endotoxin concentration. To develop sustainable raw materials for lysate reagents that do not require horseshoe crabs, three recombinant protease zymogens (factor C, derived from mammalian cells; factor B; and the proclotting enzyme derived from insect cells) were prepared using a genetic engineering technique. Recombinant cascade reagents (RCRs) were then prepared to reconstruct the reaction cascade in the amebocyte lysate reagent. The protease activity of the RCR containing recombinant factor C was much greater than that of recombinant factor C alone, indicating the efficiency of signal amplification in the cascade. Compared with the RCR containing the insect cell-derived factor C, those containing mammalian cell-derived factor C, which features different glycosylation patterns, were less susceptible to interference by the injectable drug components. The standard curve of the RCR containing mammalian cell-derived recombinant factor C had a steeper slope than the curves for those containing natural lysate reagents, suggesting a greater sensitivity to endotoxin. The present study supports the future production of recombinant reagents that do not require the use of natural resources.
Introduction
Sepsis is a serious clinical problem, the incidence of which is still increasing in the USA and worldwide. The morbidity and mortality of sepsis are reported to be nearly 660,000 cases and 17.9% in the USA in 2000, respectively. 1 In Gram-negative sepsis, endotoxin (also referred to as LPS), the major component of the outer membrane of Gram-negative bacteria, evokes serious pathologic symptoms characteristic of various forms of sepsis syndromes.2,3 The lipid moiety (lipid A) of endotoxin expresses the profound immunostimulatory capacity of the molecule. 4 Since endotoxin is also known as a pyrogenic substance in injectable drugs, preventing contamination with endotoxin is an essential element of manufacturing process control.
The bacterial endotoxin test, which uses amebocyte lysate reagents of horseshoe crab origin, is a sensitive, reproducible and simple assay to measure the endotoxin concentrations in various samples, including medical products, clinical samples of Gram-negative sepsis, 5 dialysis fluid (to evaluate its purity), 6 meat-packing (to ensure sterility), 7 milk (for the assessment of its innate immunity toward allergy prevention) 8 and samples obtained from spacecraft surfaces (to assess microbial contamination). 9 The bacterial endotoxin test is listed in pharmacopoeias as an official method and is used for the release testing of final drug products.10,11
In 1964, Levin and Bang reported that a trace amount of endotoxin coagulates Limulus hemolymph.
12
The molecular mechanism of hemolymph coagulation by endotoxin, as revealed over the last 40 years through experiments on the horseshoe crab [Tachypleus tridentatus (T. tridentatus) or Limulus polyphemus (L. polyphemus)], is based on the cascade reaction of three protease zymogens and a coagulogen,13–15 as illustrated in Figure 1(a). In this cascade, endotoxin first activates the zymogen (factor C). Activated factor C then activates factor B, which, in turn, converts the proclotting enzyme into the clotting enzyme. The clotting enzyme then cleaves coagulogen into coagulin. In horseshoe crabs, coagulin forms a gel by polymerization to coagulate their blood. Recently, we reported that, in addition to factor C, recombinant factor B binds to endotoxin.16,17 This finding suggests that in the coagulation cascade, factor B is involved not only in signal transduction from the upstream factor C, but also in endotoxin recognition. This serial activation of proteases, called the cascade reaction, is a well-conserved evolutionary phenomenon that also occurs in mammalian blood coagulation. A cascade reaction is a mechanism by which biological signals are transmitted and amplified,
18
which may increase the sensitivity to levels on the order of pg endotoxin per ml.
19
The principle of endotoxin measurement using limulus protease zymogens. (a) The coagulation cascade reaction and three techniques (gel clot, turbidimetric and chromogenic) to detect endotoxin. The dashed line indicates that endotoxin binds to factor B directly but without causing activation.
17
(b) Recombinant factor C reagent and the associated technique (fluorogenic) for endotoxin detection. (c) Schemes of the structure of the three natural protease zymogens27–29 and specific Ab epitopes (2C12, α-FCL, α-FB, and α-PCE) specific for them. H, heavy chain; L, light chain; F, full-length chain; numbers shown in parentheses, molecular weight.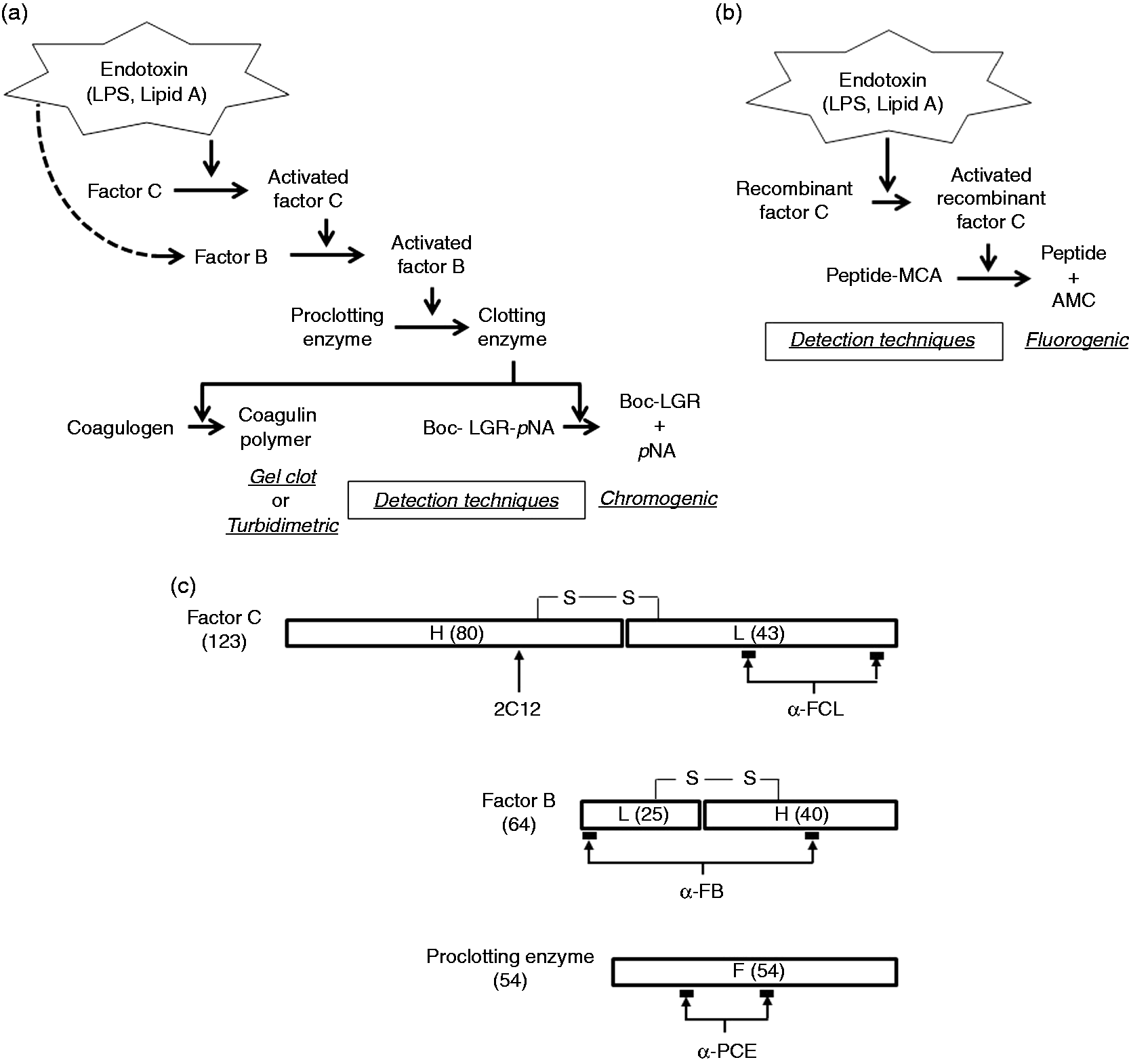
The demand for lysate reagents using horseshoe crab blood as a raw material will presumably grow with the increased use of injectable drugs, including biotechnology-based drugs and vaccines. 20 In the USA, at least 500,000 L. polyphemus living in nature are captured annually to obtain raw materials for the production of lysate reagents. The crabs are then released back to sea to prevent resource depletion. Although this series of actions (capture, blood collection and return) has been considered a causal factor of the horseshoe crab population decline based on the surveillance of the captured crabs, 21 L. polyphemus blood is still used as the sole source of raw materials for the production of lysate reagents.20,22 In South West Asia, in addition to habitat destruction, the absence or poor management of harvest regulations is considered the main cause of a decline in the number of T. tridentatus.20,23,24 Moreover, large- and commercial-scale cultivation of horseshoe crabs has never been achieved.
To bypass the unsustainable requirement for raw materials and protect the environment, recombinant factor C reagents that use the cloned factor C gene of the horseshoe crab Carcinoscorpius rotundicauda (C. rotundicauda) 25 or T. tridentatus have been marketed since 2004 as endotoxin assay reagents that do not require the use of horseshoe crabs. These reagents include recombinant factor C alone without the entire reaction cascade. To compensate for the decreased sensitivity of this system, a fluorogenic substrate that is recognized and cleaved by the activated form of recombinant factor C is used instead of a chromogenic substrate (Figure 1b). 19
In this study, to develop an endotoxin assay that does not require raw materials from horseshoe crabs, we employed a genetic engineering approach to prepare the three protease zymogens of the horseshoe crab coagulation cascade using the genes of T. tridentatus and used these zymogens in the first three steps of the cascade reaction. In addition, we evaluated three types of recombinant factor C to investigate the effects of changes in the glycosylation patterns on the reaction cascade.
Materials and methods
Materials
A United States Pharmacopeia Reference Standard Endotoxin (U.S. Pharmacopeial Convention, Rockville, MD, USA) was used in this study. Endotoxin-free plastic instruments and the Endospecy® ES-50M kit were purchased from the Seikagaku Corporation (Tokyo, Japan); the chromogenic synthetic substrates Boc-Leu-Gly-Arg(LGR)-pNA, Boc-Val-Pro-Arg(VPR)-pNA and Boc-Met-Thr-Arg(MTR)-pNA (Boc, tert-butoxycarbonyl; para-nitroanilide, pNA) were purchased from the Peptide Institute (Osaka, Japan); protease inhibitors leupeptin and pepstatin A were purchased from Merck KGaA (Darmstadt, Germany); water for injection was purchased from Otsuka Pharmaceutical (Tokyo, Japan); the eight injectable drugs were purchased from Otsuka Pharmaceutical or FUSO Pharmaceutical Industries (Osaka, Japan); the protein assay kit was purchased from Bio-Rad (Hercules, CA, USA); and all other reagents for the biochemical assays were purchased from Sigma-Aldrich Japan (Tokyo, Japan).
Abs
2C12, 26 α-FCL 17 and α-FB 17 were used as Abs against the factor C heavy (H) chain, the factor C light (L) chain and factor B, respectively. The Ab for the proclotting enzyme (α-PCE) was prepared by Scrum (Tokyo, Japan). Briefly, rabbits were immunized with two synthetic peptides, Cys-SSHVISSTQAPPETTTTE and Cys-NLYSTDDDSNPIDFA, based on the amino-acid sequence of the proclotting enzyme of T. tridentatus. The resulting antiserum was affinity-purified using antigenic peptides. Figure 1(c) illustrates the epitopes for each Ab. The specificity of these Abs was examined by Western blotting. The bands corresponding to the natural zymogens were detected under reducing or nonreducing conditions and in accordance with band sizes described in previous studies.27–29 We also verified that each Ab reacted specifically with its corresponding recombinant zymogen.
Gel electrophoresis and Western blotting
Samples containing three protease zymogens of T. tridentatus were denatured in the presence or absence of the reducing agent DTT. The resulting samples were separated by SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad). After blocking the PVDF membrane with skimmed milk, serial reactions were conducted using a primary Ab (1000–3000-fold dilutions) and a secondary Ab labeled with HRP (Dako, Glostrup, Denmark). To verify the presence of the target proteins, a luminescent substrate (Thermo Fisher Scientific, Waltham, MA, USA) was allowed to react with the secondary Ab.
Production of three protease zymogens by recombinant baculovirus
The T. tridentatus genes that encode the three protease zymogens were separately cloned into the baculovirus transfer vector pPSC8 (Protein Sciences, Meriden, CT, USA). Recombinant baculoviruses (AcNPV) obtained by homologous recombination were propagated by the infection of an Sf9 insect cell line (Novagen, Madison, WI, USA) in the logarithmic growth phase at MOI of 0.5–1.0. The culture period lasted for an additional 48–96 h. The culture supernatant was obtained by centrifugation and filtered through a polyether sulfone membrane with a pore size of 0.1 µm (Millipore, Darmstadt, Germany). The protein concentrations in the resulting filtrates were measured using the Bio-Rad protein assay kit.
Production of three protease zymogens using the Sf9 cell line using a stable expression system
The three respective genes of T. tridentatus encoding the protease zymogens were cloned into the vector pIZ-V5 (Invitrogen, Carlsbad, CA, USA) for stable expression in insect cells. The three resulting expression plasmids were separately transfected into Sf9 cells using Cellfectin® II Reagent (Invitrogen). Using the Zeocin-tolerant marker gene on the vector as an index, the cell lines in which the expression plasmids were integrated into genomic DNA were obtained using a colony-formation method. 30 To screen for high-expressing cell lines containing the target zymogens, the expression levels of the recombinant proteins in the culture supernatant were assessed by Western blotting. The cell lines were subsequently subjected to suspension culture. The culture supernatant was obtained by centrifugation and filtered through a PVDF membrane with a pore size of 0.22 µm (Millipore). The protein concentrations in the resulting filtrates were measured.
Production of factor C using the CHO DG44 cell line using a stable expression system
The T. tridentatus gene encoding factor C was cloned into the mammalian expression vector pCI-neo (Promega, Madison, WI, USA). The resulting factor C expression plasmid and an expression plasmid encoding a dihydrofolate reductase were simultaneously transfected into a CHO DG44 cell line using the Lipofectin® reagent (Invitrogen). The gene-integrated cell lines were selected in medium containing 1 mg/ml geneticin. Subsequently, the methotrexate concentrations in the culture medium were increased in a stepwise manner, resulting in the construction of factor C gene-amplified cell lines. Using the RNA and protein expression levels and the protease activity of factor C as indices, the cell lines that highly expressed factor C were selected using a limiting dilution approach. The relevant cell lines were conditioned to and floated in a chemically defined medium. The culture supernatant was collected by centrifugation and filtered using a PVDF membrane with a pore size of 0.22 µm (Millipore). The protein concentration in the resulting filtrate was measured.
Production of factor C using the HEK293 cell line using a transient expression system
The T. tridentatus gene encoding factor C was cloned into a mammalian expression vector, pCA7, and the factor C gene was expressed by transient transfection in HEK293 cells. 16 The culture supernatant was obtained by centrifugation and filtered through a PVDF membrane with a pore size of 0.22 µm (Millipore). The protein concentration in the resulting filtrate was determined.
Purification of four types of factor C
Each of the three types of culture supernatants or a T. tridentatus amebocyte lysate (TAL), which contained any one of four types of factor C, was applied to a dextran sulfate-Sepharose CL-6B (GE Healthcare, Uppsala, Sweden) column, and fractions eluted with 20 mM Tris hydrochloride buffer (pH 8.0) containing 450 mM NaCl were collected.29,31 Fractions containing factor C were identified by Western blotting using an anti-factor C Ab and by determining the factor C activity. Subsequently, the fraction containing the highest amount of factor C was applied to the CL-6B sepharose column for gel filtration. 32 The protein concentrations in the eluted samples were determined.
Quantitation of bacterial endotoxins using recombinant zymogens
In the presence of 100 mM Tris hydrochloride buffer solution (pH 8.0), a synthetic substrate and recombinant zymogens were diluted with water for injection to obtain protein concentrations of 120–240 µg/ml under cooling conditions, and the resulting solution was used as the enzyme solution. Water for injection or an injectable drug containing endotoxin was prepared, and 50-μl aliquots were dispensed into a 96-well microplate. The enzyme solution (50 μl) was then added to the plate to achieve a total volume of 100 μl. The plate was placed in a microplate reader (Wellreader MP-96; Seikagaku Corporation) at 37℃, and the increase in the absorbance at 405 nm (reference wavelength: 492 nm) was measured at 15-s intervals for 30 or 60 min. A dedicated software program was used to analyze the average change in absorbance per min to evaluate protease activity.
Treatment of factor C with glycopeptidase F
The four types of factor C were treated with glycopeptidase F (Takara Bio, Kyoto, Japan). After 14 h incubation in the presence of DTT at 37℃ according to the manufacturer's instructions, the change in the size of factor C was examined by Western blotting with 2C12 and α-FCL Abs.
Analysis of the lectin-binding ability of factor C
Dot blots were prepared by spotting the four types of purified factor C and fetuin onto a nitrocellulose membrane (Bio-Rad). After blocking the blots with BSA, serial reactions were initiated with biotinylated lectin (J-Oil Mills, Tokyo, Japan) and HRP-labeled streptavidin (Thermo Scientific) to verify the presence of target proteins using the luminescence caused by HRP substrate decomposition.
Results
Transient expression of three protease zymogens by recombinant baculovirus
Ding et al., based on their studies using C. rotundicauda gene, reported the inability to obtain recombinant factor C that can be activated by endotoxin using yeast,33–35 S2 cells
36
and COS-1 cells
37
as hosts. However, in a different study that used an Sf9-baculovirus system,
38
the investigators successfully obtained recombinant factor C that could be activated with endotoxin. We produced three recombinant protease zymogens by expressing each of the respective T. tridentatus genes using an Sf9-baculovirus system. Western blotting demonstrated that target recombinant zymogens were secreted into the culture supernatants after infection with recombinant viruses. The bands corresponding to factor C and the proclotting enzyme were verified 48 h after infection. However, after 72 h or more, the bands observed at 48 h disappeared, and several smaller bands appeared (Figure 2a). We confirmed reductions in protease activity in accordance with these changes in band size (data not shown), suggesting that the target recombinant zymogens had been degraded. The degradation was not completely suppressed by the addition of protease inhibitors (leupeptin and pepstatin A that are effective on baculovirus-derived proteases)
39
to the culture solution. These results demonstrated that the target zymogens were secreted extracellularly after translation and were degraded in this baculovirus system in a time-dependent manner.
Expression of three protease zymogens using transient and stable expression systems. (a) Western blotting showing the generation of protease zymogens by recombinant baculoviruses in the absence of DTT. The numbers in the lanes represent the culture time after viral infection. (b) Western blotting showing the generation of factor C using a stable expression system in Sf9 cells. Control: Sf9-baculovirus system. The numerals in the lanes represent the culture time after passage. (c) Western blotting showing the generation of factor B using a stable expression system in Sf9 cells. The numerals in the lanes represent the culture time after passage. (d) Western blotting showing the generation of the proclotting enzyme using a stable expression system in Sf9 cells. The numerals in the lanes represent the volume (μl) of the sample applied to the gel. F, full-length chain.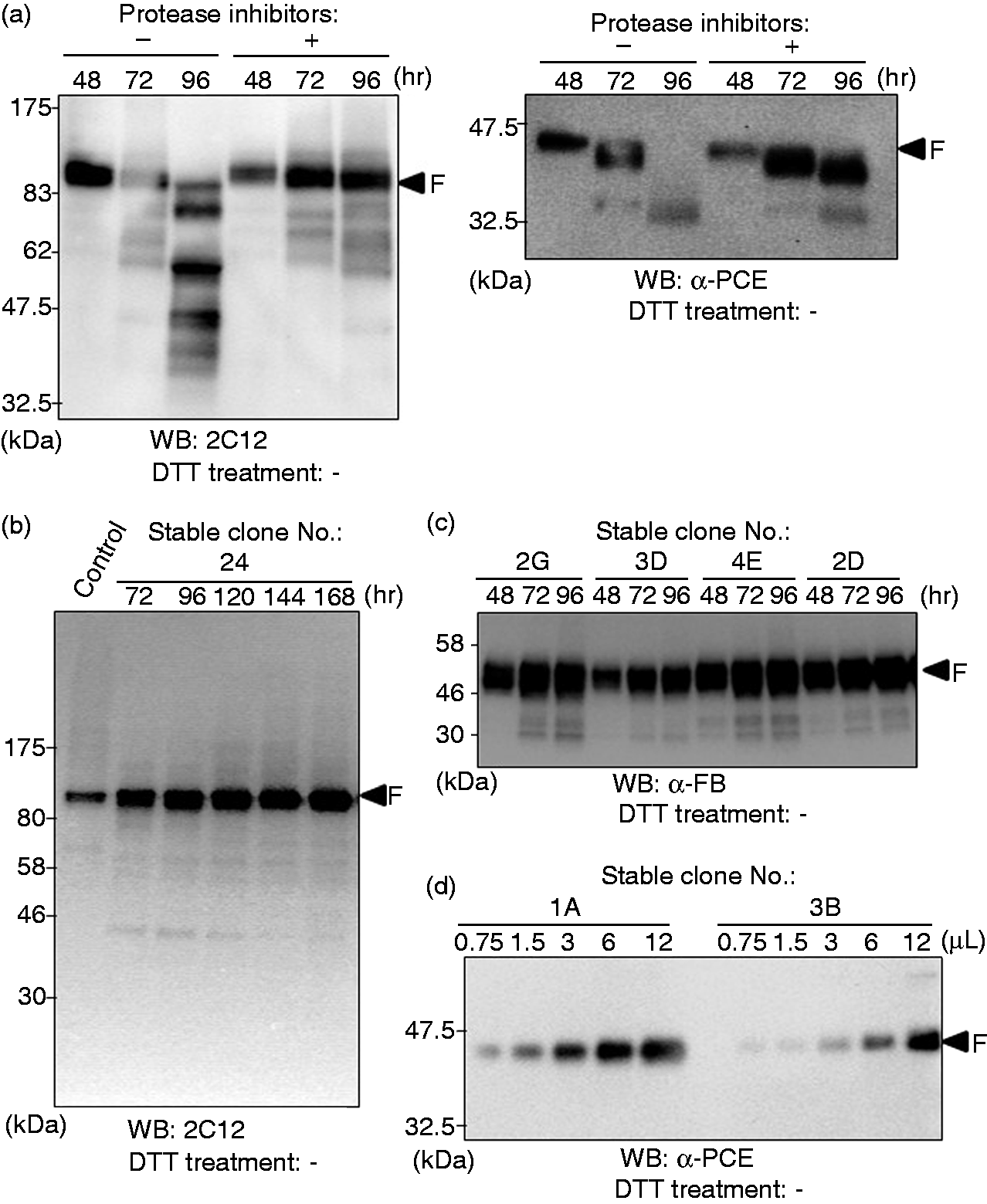
Stable expression of three protease zymogens in the Sf9 cell lines
To further confirm that the degradation of the recombinant zymogens was caused by baculovirus-derived proteolytic enzymes, a baculovirus-free stable expression system in which the target genes of zymogens are integrated directly into the genomic DNA of Sf9 cells was used to produce the recombinant zymogens.
We cultured the candidate cell lines, which were cloned using the colony-formation method, and performed Western blotting of the culture supernatant. Unlike the baculovirus method, the target zymogens were not degraded over time in culture. The yield of factor C protein in the culture supernatant was higher than that obtained 48 h after viral infection (Figure 2b). The yields of secreted recombinant factor B (Figure 2c) and the proclotting enzyme (Figure 2d) clearly differed among several candidate cell lines. Therefore, we selected the cell lines with optimal growth and survival rates in addition to sufficient zymogen expression levels, and thus we succeeded in obtaining three intact recombinant zymogens.
Reconstruction of the first three steps of the coagulation cascade reaction
The first three steps of the coagulation cascade reaction had been successfully reconstructed using three natural factors purified from TAL.
28
To confirm whether the three recombinant zymogens can similarly reconstruct the reaction steps, we used seven combinations of the three recombinant zymogens in the presence of a chromogenic synthetic substrate that is cleaved by the clotting enzyme (i.e. Boc-LGR-pNA). We subsequently verified that the clotting enzyme activity was induced by endotoxin only when all three recombinant zymogens were present in a set (Figure 3a). When the three natural protease zymogens in TAL are activated serially in the coagulation cascade reaction, each protease zymogen undergoes limited proteolysis.27–29 Indeed, Western blotting verified that the three recombinant zymogens had undergone limited proteolysis after contact with endotoxin in the reconstructed reaction (Figure 3b). To assess whether the protease activity induced by factor C in the cascade reaction was serially amplified, we measured the time-dependent changes in absorbance at 405 nm for the three combinations of recombinant zymogens and their specific substrates, Boc-VPR-pNA
40
, Boc-MTR-pNA
28
and Boc-LGR- pNA, after the addition of endotoxin (Figure 3c). In combination A, consisting of factor C and Boc-VPR-pNA, the protease activity hardly increased. In combination B, consisting of factor C, factor B and Boc-MTR-pNA, the protease activity increased to a slightly greater extent than that observed for combination A. In combination C, consisting of factor C, factor B, the proclotting enzyme and Boc-LGR-pNA, the protease activity increased rapidly and reached a plateau after approximately 15 min of incubation. Under the conditions used in the present study, we confirmed the occurrence of signal amplification because the protease activity was greatly enhanced in the presence of the three recombinant zymogens compared with that of recombinant factor C alone.
Reconstruction of the coagulation cascade reaction using three recombinant zymogens. (a) Protease activity in the different combinations of the three recombinant zymogens. The protease activity in milliabsorbance units (mAbs)/min is defined as the average rate of change in milliabsorbance units per unit time over 60-min incubation. Error bars represent the SD of three experiments. (b) Western blotting showing limited proteolysis after contact with endotoxin in the reconstructed first three steps of the coagulation cascade. (c) Time-dependent changes (0–60 min) in protease activity with different combinations of the three recombinant zymogens and their specific substrates. EU, endotoxin unit; FC, factor C; FB, factor B; PCE, the proclotting enzyme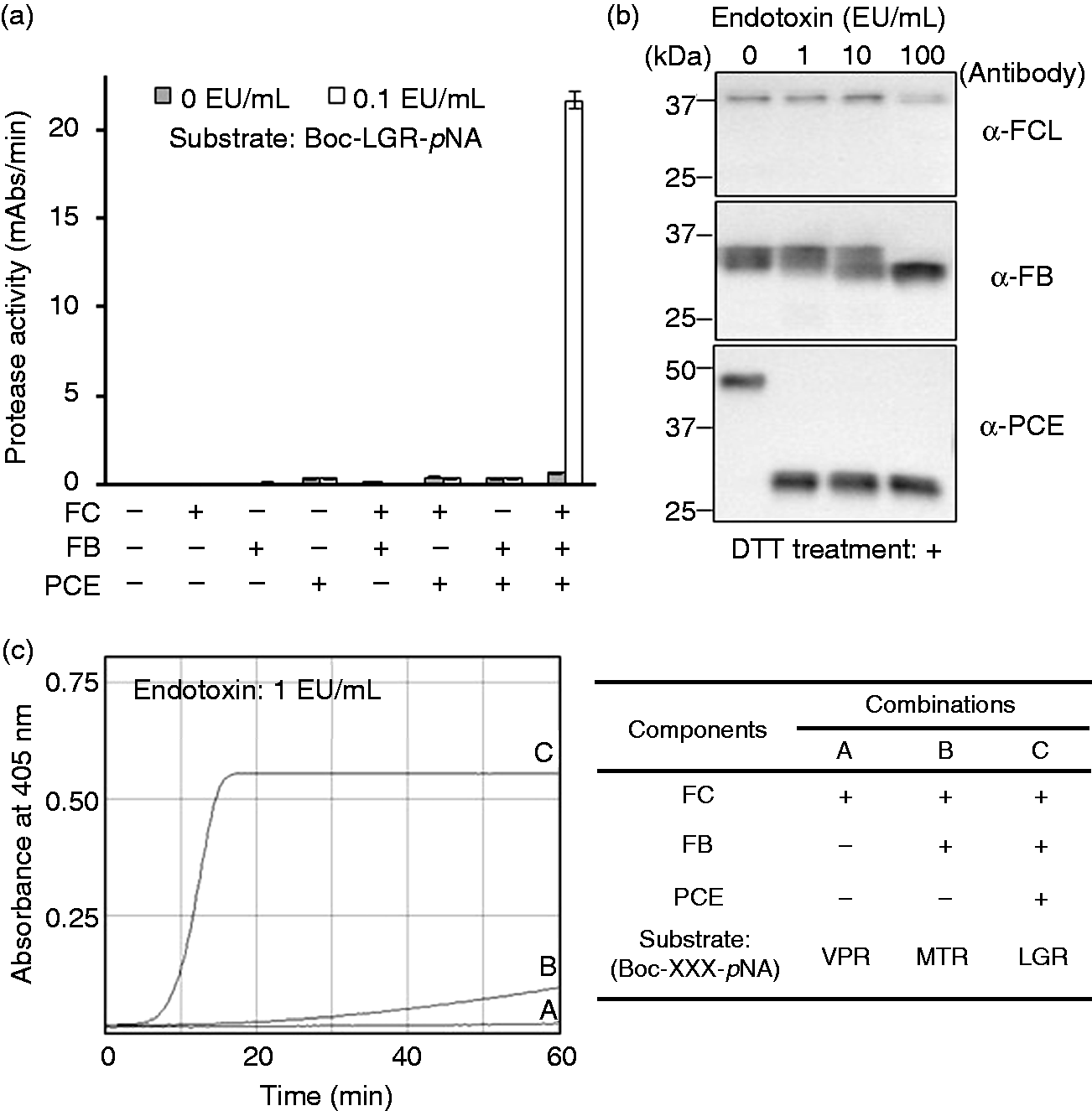
Effects of post-translational glycosylation in transfected cells on the apparent molecular size of three types of recombinant factor C
The three recombinant zymogens exhibited differences in migration on Western blots compared with that of natural zymogen (Figure 4a). To examine the effects of post-translational modifications in the transfected cells on the apparent molecular size of factor C, three types of recombinant factor C were prepared using two mammalian cell lines and the Sf9 insect cell line.
The glycosylation pattern analyses of the four types of purified factor C. (a) Western blotting using Abs (2C12, α-FCL, α-FB, and α-PCE) against natural and three recombinant zymogens in the presence (+) or absence (-) of DTT. Lane 1: partially purified natural products containing factors C and B, 2: partially purified natural products containing the proclotting enzyme, 3: CHO DG44 culture supernatant containing recombinant factor C, 4: Sf9 culture supernatant containing recombinant factor B, and 5: Sf9 culture supernatant containing recombinant proclotting enzyme. (b) Coomassie brilliant blue (CBB) staining of purified factor C in the presence (+) or absence (–) of DTT. (c) Western blotting of anti-factor C H- and L-chain Abs, i.e. 2C12 and α-FCL, respectively, in the presence (+) or absence (–) of DTT. (d) Western blots of anti-factor C H- and L-chain Abs (i.e. 2C12 and α-FCL, respectively) with or without treatment with glycopeptidase F and in the presence of DTT. (e) Lectin-binding analysis of purified factor C using fetuin as the control. Lane 1: factor C from T. tridentatus amoebocyte lysate; 2: recombinant factor C from CHO DG44 cells; 3: recombinant factor C from HEK293 cells; and 4: recombinant factor C from Sf9 cells. H, heavy chain; L, light chain; F, full-length chain.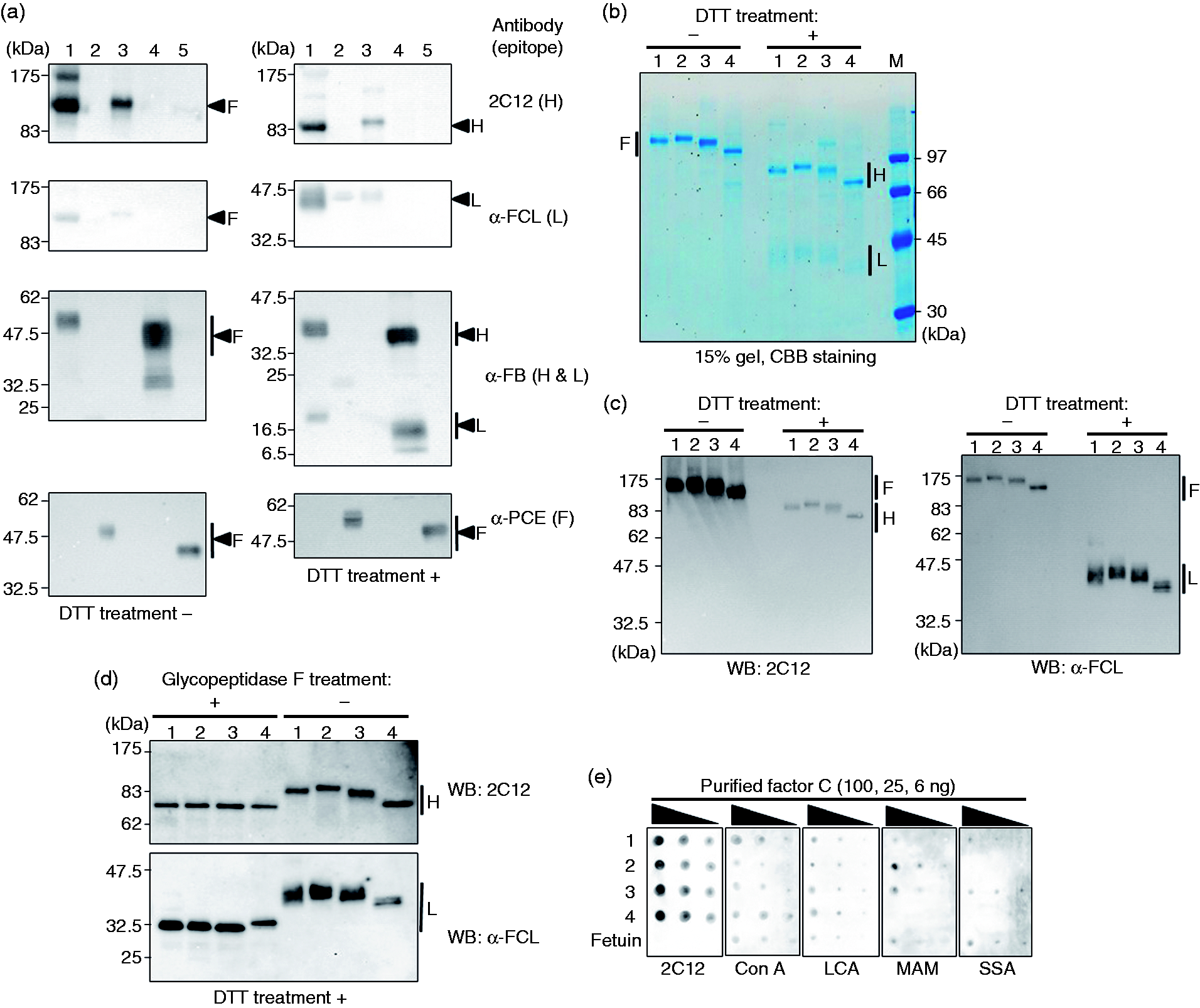
Four types of purified factor C, including the natural form (CTAL) and the three recombinant forms derived from the cell lines CHO DG44 (CCHO), HEK293 (CHEK) and Sf9 (CSF), were obtained by column chromatography and subjected to SDS-PAGE. Subsequently, the apparent molecular masses of these types of factor C were compared using Coomassie brilliant blue (CBB) staining and Western blotting (Figure 4b and 4c, respectively). The largest recombinant factor C protein (H + L chains) under non-reducing conditions was CCHO, followed by CTAL, CHEK and CSF. Under reducing conditions, these L chains were less prone to staining with CBB 40 but can be identified with α-FCL. Both the H and L chains exhibited the above-mentioned trends in differences in molecular mass.
Glycosylation is a typical post-translational protein modification, and there are five potential N-glycosylation sites (three and two sites for the H and L chains, respectively) in the amino-acid sequence of factor C. 41 To assess whether differences in the sizes of CTAL, CCHO, CHEK and CSF (shown in Figure 4b and c) were due to N-glycosylation, we removed N-glycans with glycopeptidase F (Figure 4d). The H chains of CTAL, CCHO, CHEK and CSF displayed reduced sizes compared with those found prior to the enzyme treatment, respectively, and were equivalent in size among the treated samples. However, the enzyme-treated L chains of CTAL, CCHO, CHEK and CSF were also smaller, and that of CSF was slightly larger than those of CTAL, CCHO and CHEK. These data suggested the presence of an N-glycan uncleavable by glycopeptidase F 42 and/or causes other than N-glycosylation.
Glycans are transferred to the asparagine residues of mammalian-cell proteins in a stepwise manner via N-glycosylation, and hybrid or complex sugar chains containing terminal sialic acids are ultimately formed. However, pauci-Man- or high Man-type sugar chains are often formed in insect cells because of the lack of sialyltransferase. 43 To examine the differences in the glycosylation pattern of each factor C, we conducted lectin-binding analysis of the purified molecules (Figure 4e). Consistency in the amounts of each purified factor C retained on the nitrocellulose membrane was confirmed using the 2C12 Ab. Canavalia ensiformis agglutinin (Con A), an α-Man-specific lectin, and Lens culinaris agglutinin (LCA), a Fuc-specific lectin, both bound to fetuin, which was used as a control, and to CTAL, CCHO, CHEK and CSF. In contrast, Maackia amurensis agglutinin (MAM), which is specific for terminal sialic acids with α-(2→3) binding, bound strongly to CCHO and CHEK in decreasing order, slightly to CTAL and fetuin, and not to CSF. Sambucus sieboldiana agglutinin (SSA), which is specific for terminal sialic acids with α-(2→6) binding, bound strongly to CTAL, CHEK and fetuin but did not bind to CCHO or CSF. These results suggested that N-glycosylation containing terminal sialic acids were present in CCHO and CHEK prepared using mammalian cells but not in CSF prepared using insect cells.
Effect of the differences in the glycosylation pattern of factor C on the reconstructed cascade reaction
Lysate reagents are used extensively for the quality control of injectable drugs. However, the endotoxin-triggered reaction in these reagents is inhibited by certain components of such drugs. Therefore, inhibitory components in injectable drugs must be removed or those drugs must be diluted to a level at which such an inhibitory influence is negligible.
Recombinant proteins, even with the same amino-acid sequences, are well known to exhibit different activity and stability depending on their different post-translational modifications.
44
To examine the effects of the different glycosylation patterns in CTAL, CCHO, CHEK and CSF on the susceptibility to interference by the components of injectable drugs, we prepared four different recombinant cascade reagents (RCRs) by adding each of the four types of factor C to a mixture of the synthetic substrate (Boc-LGR-pNA), recombinant factor B and the proclotting enzyme (Figure 5a). First, we determined the conditions under which the four RCRs exhibited an equivalent ability to detect endotoxin in water for injection (Figure 5b). Subsequently, we compared the levels of the reaction interference for four RCRs using eight commercially available injectable drugs that had been intentionally contaminated with endotoxin. The residual protease activity, expressed as the activity relative to the activity found in water for injection (which was defined as 100%), differed depending on the type of injectable drug examined. In particular, RCRs containing CCHO and CHEK (mammalian cells) were less susceptible to interference than those containing CSF and CTAL; furthermore, CCHO exhibited the greatest residual protease activity, except when sodium citrate was used as the injectable drug (Figure 5c). These results indicate that CCHO is a preferred component of RCRs because it is less susceptible to interference.
Comparison of the four types of factor C with different glycosylation patterns regarding the susceptibility to interference by injectable drugs. (a) Procedures for bacterial endotoxin quantification using the three recombinant zymogens. (b) Protease activity of RCRs using the four types of factor C vs. water for injection. The protease activity in mAbs/min was defined as the average change rate in milliabsorbance units per unit time over 30 min incubation. (c) Injectable drug-mediated inhibition of the protease activity of RCRs using the four types of factor C. Residual protease activity: activity relative to that obtained when using water for injection, defined as 100%. Conc.: labeled concentration of a purchased injectable drug; dilution: dilution factor used when measuring a sample with RCRs. (d) Standard curves of RCR and a natural lysate reagent (ES-50M). Error bars represent the SDs from three experiments. EU, endotoxin unit; CTAL, purified factor C from Tachypleus tridentatus; CCHO, recombinant factor C from CHO DG44; CHEK, recombinant factor C from HEK293; CSF, recombinant factor C from Sf9; RCR, recombinant cascade reagent.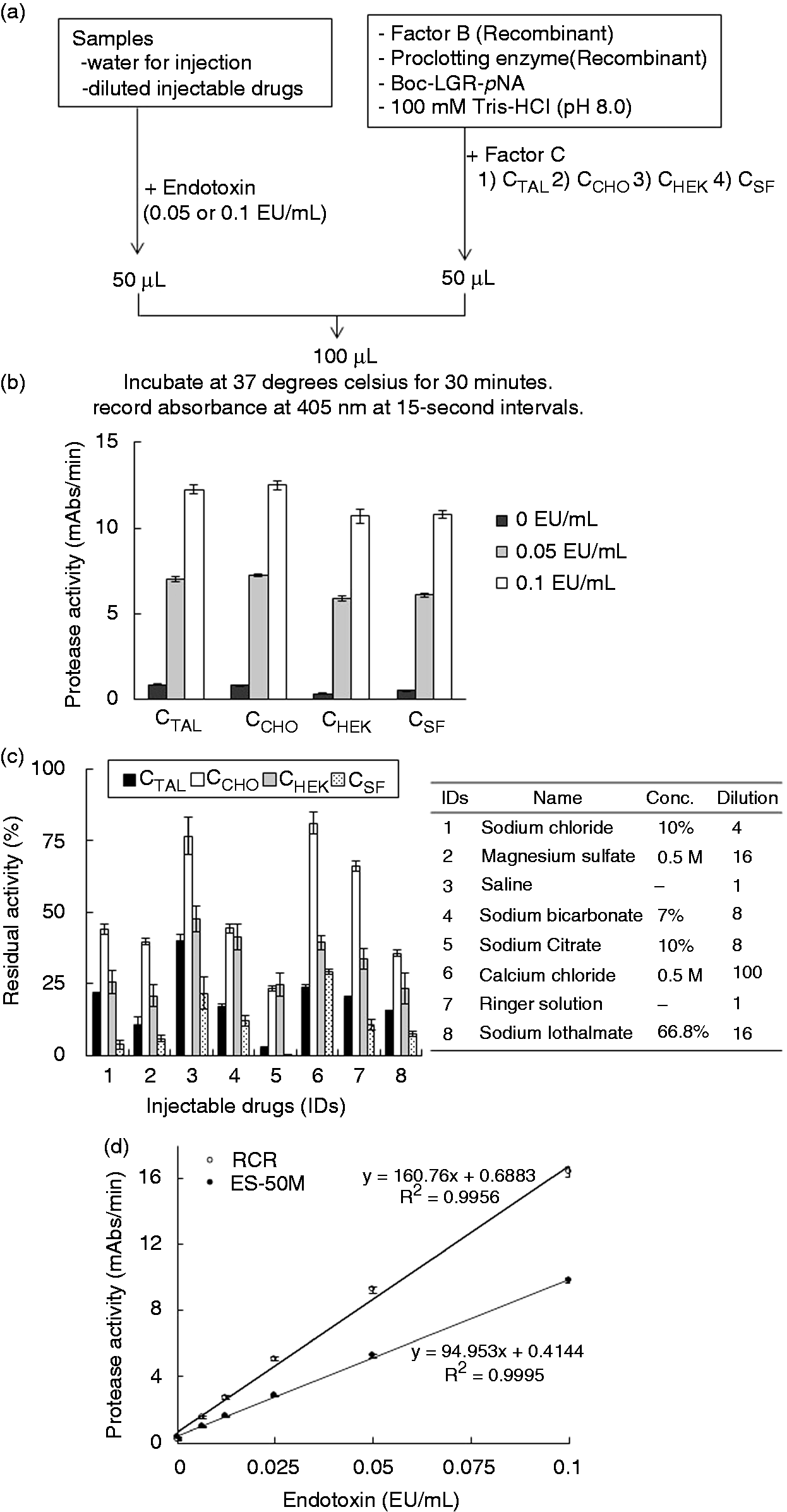
We prepared an RCR using CCHO, Sf9-derived factor B and Sf9-derived proclotting enzyme as raw materials and compared the prepared RCR with a commercialized natural lysate reagent (Endospecy ES-50M) in terms of sensitivity to various concentrations of endotoxin in water. The RCR displayed greater sensitivity to endotoxin than did the natural lysate reagent, as evidenced by the steeper slope of the RCR standard curve (Figure 5d).
Discussion
We report the first RCR produced by the in vitro reconstruction of the first three steps of the Limulus coagulation cascade reaction, consisting of three recombinant zymogens. Furthermore, we demonstrated that this cascade was efficient for signal amplification and allows for highly sensitive endotoxin detection. The RCR may be considered a promising substitute for natural lysate reagents because it requires only the tools and instruments that are normally used with the natural lysate reagent, while recombinant factor C reagent requires a special fluorescence microplate reader (Figure 1b). 19
We propose that our genetic engineering approach provides the following theoretical advantages: (1) no competition occurs between the synthetic substrate and naturally existing substrate, coagulogen, both of which are cleaved by the clotting enzyme. (2) The RCR may offer potent protease activity because it lacks the protease inhibitors that are present in the horseshoe crab amebocyte, including serpin-1 for activated factor C and serpin-2 for the clotting enzyme. 15 In fact, the RCR exhibits more potent protease activity than the natural lysate reagent (Figure 5d). (3) β-Glucan-induced false positivity does not occur because the RCR does not contain factor G, which is a β-glucan-sensitive zymogen and whose activated form activates the proclotting enzyme.31,45 (4) Batch-to-batch fluctuations in the sensitivity of natural lysates, 38 which may be caused by individual differences in horseshoe crabs and their living environments, should be reduced in the RCR system.
In this study, we found that CCHO is the more promising form of factor C than CSF owing to its reduced susceptibility to interference by components of the examined injectable drugs. Unlike insect cells, mammalian cells undergo the addition of terminal sialic acids during N-glycosylation.43,44 We presume that such additions may contribute to less susceptibility of CCHO, but further studies are necessary here.
Liquid raw materials are currently combined to create the RCR. However, we emphasize the need to prepare a lyophilized RCR similar to the natural lysate reagents due to its storage and transport stability. Furthermore, using natural lysate reagents as controls, we intend to examine in detail the analytical characteristics of the lyophilized RCR and its susceptibility to the components of the injectable drugs listed in the pharmacopoeias, in addition to its application in clinical use.
In conclusion, the present study indicates the potential use of recombinant reagents for the bacterial endotoxin test to overcome the requirement for natural resources.
Footnotes
Acknowledgments
The authors thank Satoshi Sakima, MD, for his gracious review of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the joint research fund of the Seikagaku Corporation to SK.