
Abstract
Pro-resolving, docosahexaenoic acid-derived mediators have recently emerged as important potential therapeutic agents for the amelioration of complications arising from inflammation, such as vascular disease, asthma, acute lung injury and colitis. While resolvin D1 (RVD1), resolvin D2 (RVD2) and maresin 1 (MaR1) are established pro-resolvins, their mechanisms of action remain unclear. Here we show that, in LPS-stimulated primary human monocytes, RVD1, RVD2 and MaR1 each suppress the release of pro-inflammatory cytokines (TNF, IL-1β, IL-8) and the innate/adaptive bridging cytokine, IL-12 p40, while simultaneously augmenting the production of the anti-inflammatory cytokine, IL-10. Such resolving activity is accompanied by the increased phosphorylation (enhanced anti-inflammatory state) of glycogen synthase kinase 3β (GSK3β) along with increased phosphorylation (activation) of Akt, SGK1 and CREB but not MAPK-related molecules. Gain and loss of function experiments confirm a key role for GSK3β and CREB in the anti-inflammatory actions of resolvins. These results suggest that induction of the GSK3β anti-inflammatory axis is a common mechanism of action for RVD1, RVD2 and MaR1.
Introduction
There are multiple potentially convergent endogenous pathways that serve to limit inflammation and promote its resolution, thereby protecting the host from collateral damage. Such innate braking systems include the α7 acetylcholine receptor-dependent cholinergic; glycogen synthase kinase 3β (GSK3β); serum and glucocorticoid-regulated kinase 1 (SGK1); and Wnt anti-inflammatory signaling axes.1–5 The identification of omega-3 polyunsaturated fatty acid-derived resolving mediators, including the docosahexaenoate metabolites resolvin D1 (7S,8R,17S-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid; RVD1), resolvin D2 (7S,16R,17S-trihydroxy-4Z,8E,10Z,12E,14E,19Z-docosahexaenoic acid; RVD2) and maresin-1 (7R,14S-dihydroxy-4Z,8E,10E,12Z,16Z,19Z-docosahexaenoic acid; MaR1), have added to the emergent understanding that the resolution of inflammation is highly dynamic.6–9
In vivo, RVD1, RVD2 and/or MaR1 have been shown to suppress pro-inflammatory cytokine production during multiple inflammatory conditions with protective consequences, including experimental uveitis, 10 hepatic ischemia/reperfusion injury, 11 peritonitis, 12 periodontitis, 13 acute lung injury,14–18 colitis,19,20 endometriosis 21 and endotoxic shock. 22 Resolvins have also been reported to abet caspase-dependent neutrophil apoptosis,14,23 to augment efferocytosis24,25 and to promote the de novo generation of regulatory T cells. 26 All such processes are associated with the resolution of inflammation.
In vitro, RVD1, RVD2 and/or MaR1 suppress pro-inflammatory cytokine release in several innate immune cell types that have been exposed to variant inflammatory mediators, such as bronchial epithelial cells,27,28 corneal epithelial cells, 29 macrophages 30 and gingival fibroblasts. 31 Furthermore, resolving mediators, particularly RVD1 and MaR1, have been shown to suppress NF-κB activation in cells such as microglia,32,33 smooth muscle cells 34 and vascular endothelial cells, 35 as well as in several inflammatory disease animal models, including acute lung injury,15,18,36 colitis 19 and sepsis. 37 However, the events leading to inhibition of NF-κB signaling have yet to be fully elucidated.
GSK3β is constitutively active and promotes inflammation in the context of TLR engagement. However, phosphorylation of GSK3β at Ser9 results in inhibition of the production of pro-inflammatory cytokines.1,2,38 Zhang et al. 11 recently suggested that RVD1 suppression of pro-inflammatory cytokines and attenuation of hepatic ischemia/reperfusion injury is mediated, at least in part, by a mechanism involving enhanced phosphorylation of Akt. Several other groups have also reported that PI3K-Akt-related signaling events are key to the protective efficacy of RVD1 protection against inflammatory damage.16,39,40 Further, resolvin E1 (5S,12R,18R)-trihydroxy-6Z,8E,10E,14Z,16E-eicosapentaenoic acid; RVE1) and lipoxin A4 (5S,6R,15S-trihydroxy-7E,9E,11Z,13E-eicosatetraenoic acid; LXA4) have both been reported to influence the activation state of Akt.41,42 PI3K is an activator of Akt, while Akt is an established kinase inactivator of the central mediator of inflammation, GSK3β.1,2,38 Therefore, we hypothesized that the pro-resolving activities of RVD1, RVD2 and MaR1 may involve the inactivation of GSK3β and thus augmentation of GSK3β anti-inflammatory axis. We set out to test this hypothesis in LPS-stimulated primary human monocytes.
Materials and methods
Materials
Monocyte isolation kits were from Miltenyi Biotec (Auburn, CA, USA). RPMI Complete (RPMI 1640 medium supplemented with FBS, 2-mercaptoethanol, sodium pyruvate,
Preparation of human monocytes
Primary human monocytes were purified from anonymized, citrated whole blood by indirect magnetic monocyte isolation, as approved by the University of Louisville, Institutional Review Board (12.0346). This procedure routinely results in >95% pure CD14+ cells, as shown by flow cytometry. Human monocytes were cultured at 37℃ and 5% CO2 atmosphere, in complete RPMI plus or minus stimulating agents, as described below. Monocyte viability was evaluated by trypan blue exclusion.
Cytokine release by human monocytes
Primary human monocytes (200,000 cells/well) were stimulated or not with LPS and/or MaR1, RVD1 or RVD2, as described in the figure legends. Ethanol solvent controls, equivalent to the highest solvent concentration in the resolving stimulations, were included. MaR1, RVD1 or RVD2 was added 30 min prior to TLR4 stimulation with LPS. Cell-free supernatants were harvested at 20 h and assayed for cytokine levels by ELISA, according to the manufacturer’s instructions.
Expression of GSK3β pathway molecules
Levels of Ser536 phosphorylated NF-κB p65, Ser9 phosphorylated GSK3β, as well as Ser133 phosphorylated CREB, Ser422 phosphorylated SGK-1, pSYK, pERK1/2, pSAPK/JNK, p38 MAPK, β-actin and GAPDH were determined by Western blot using whole-cell lysates (10 µg protein). Images were visualized by enhanced chemiluminescence and acquired using the ImageQuant LAS 4000 system (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). Expression levels are presented as mean intensity ratios relative to a housekeeping gene, as determined by densitometry using Image J software (National Institutes of Health, Bethesda, MD, USA).
Pharmacological inhibition of GSK3β pathway
The contribution of GSK3β to the inhibition of TLR4-induced cytokine induction in human monocytes was further explored through the use of the pharmaceutical inhibitor, SB216763. Monocytes were pre-treated with SB216763 for 2 h prior to LPS (1 µg/ml) stimulation with or without an additional pre-LPS stimulation with MaR1. DMSO (0.05%) and ethanol (as described above) solvent controls were included. Cell-free supernatants were assayed for cytokine levels by ELISA 20 h after the addition of LPS.
Transfection of GSK3β(S9A) and siRNA
Transfection of monocytes with non-targeting control siRNA and siRNA-CREB, as well as plasmids pcDNA3-GSK3β(S9A) and pcDNA3 (empty vector control) was achieved by electroporation using Lonza Nucleofector technology (Allendale, NJ, USA), following the manufacturer’s protocol. Transfected monocytes were stimulated with LPS on d 3 post transfection. Cell-free supernatants were assayed for IL-8 or IL-10 by ELISA 20 h after the addition of LPS.
Statistical analyses
Statistical significance between groups was evaluated by the analysis of variance and the Tukey multiple comparison test using the InStat program (GraphPad, San Diego, CA, USA). Differences between groups were considered significant at the level of P ≤ 0.05. Some Western blot-related data refer to representative blots. All other data are presented as the arithmetic mean ± SD of three biological replicates.
Results
Resolvin D1, resolvin D2 and maresin-1 suppress LPS-induced pro-inflammatory cytokines in primary human monocytes
Initial experiments established that resolvins were highly efficient inhibitors of the release of IL-8, selected as a typical LPS-induced cytokine, from TLR4-engaged human monocytes across a broad range of concentrations (Figure 1).
Inhibition of LPS-induced IL-8 by variant resolvin doses. Twenty h cell-free supernatants were collected from primary human monocytes stimulated with or without LPS (1 µg/ml) in the presence or absence of (a) MaR1 (0–5 μM), (b) RVD1 (0–60 nM) or (c) RVD2 (0–2 μM). The production of IL-8 was determined by ELISA. Data represent the arithmetic mean ± SD of three biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001.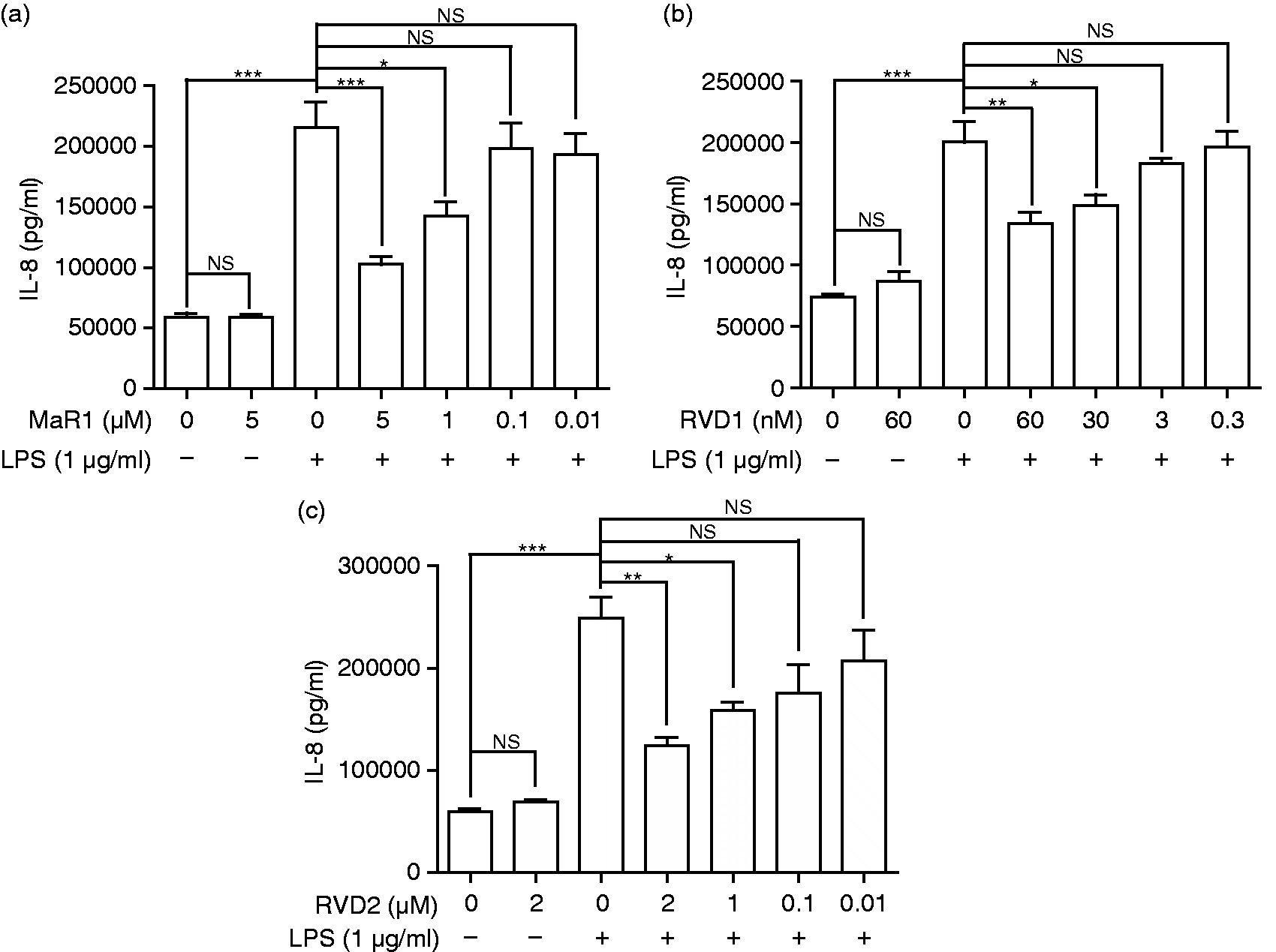
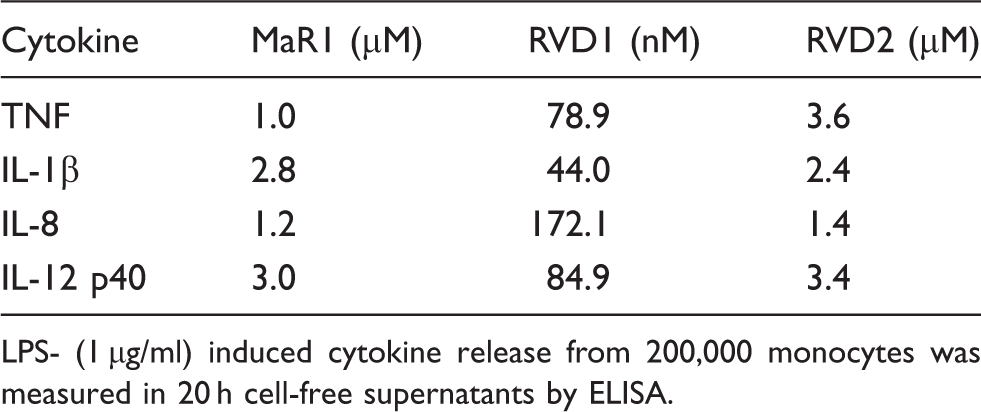
Subsequently, the ability of RVD1, RVD2 and MaR1 to inhibit inflammation-inducing cytokines (IL-1β, TNF; all P < 0.05) and the p40 sub-unit of the innate-adaptive bridging cytokine, IL-12 (all P < 0.05), was determined (Figure 2). The IC50 values for suppression of TNF, IL1β, IL-8 and IL-12 p40 are presented in Table 1. Resolvins alone did not influence pro-inflammatory cytokine secretion in innate cells (Figures 1 and 2). Furthermore, resolvins had no significant influence on monocyte viability at the concentrations employed, as determined by trypan blue exclusion.
MaR1, RVD1 and RVD2 each inhibit LPS-induced pro-inflammatory cytokines [(a) IL-1β, (b) TNF and (c) IL-12 p40]. Twenty h cell-free supernatants were collected from primary human monocytes stimulated with or without LPS (1 µg/ml) in the presence or absence of MaR1 (1 μM), RVD1 (30 nM) or RVD2 (1 μM). The production of pro-inflammatory cytokines was determined by ELISA. Data represent the arithmetic mean ± SD of three biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001. Resolving mediator concentration required to inhibit 50% cytokine release (IC50) from TLR4-engaged primary innate cells. LPS- (1 µg/ml) induced cytokine release from 200,000 monocytes was measured in 20 h cell-free supernatants by ELISA.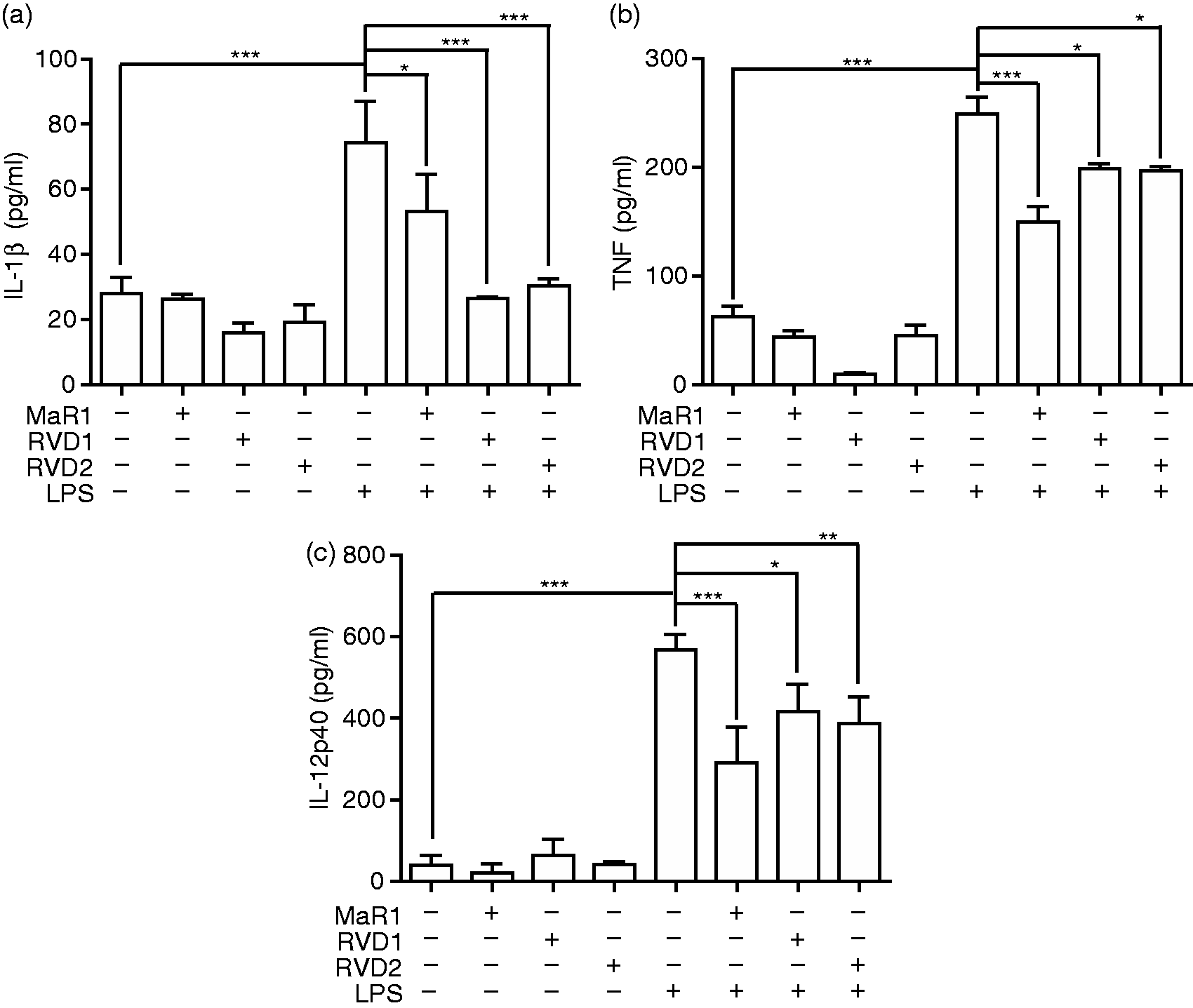
Resolvin D1, resolvin D2 and maresin-1 augment LPS-induced anti-inflammatory IL-10 release
Concomitant with the suppression of pro-inflammatory cytokines, RVD1, RVD2 and MaR1 each augmented the release of the anti-inflammatory cytokine, IL-10 (all P < 0.05) from TLR4-engaged innate cells, as shown in Figure 3. Resolvins alone did not influence IL-10 production in human monocytes (Figure 3).
MaR1, RVD1 and RVD2 each augment LPS-induced IL-10 release. Twenty h cell-free supernatants were collected from primary human monocytes stimulated with or without LPS (1 µg/ml) in the presence or absence of MaR1 (1 μM), RVD1 (30 nM) or RVD2 (1 μM). The production of IL-10 was determined by ELISA. Data represent the arithmetic mean ± SD of three biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001.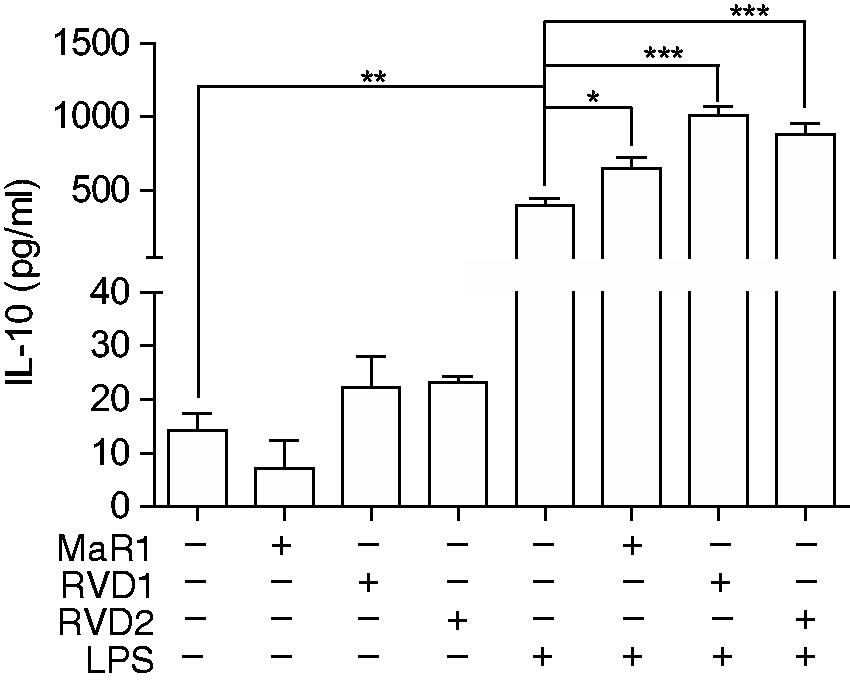
Resolvin D1, resolvin D2 and maresin-1 phospho-inactivate GSK3β while activating CREB in LPS-stimulated innate cells
As can be seen in Figure 4, RVD1, RVD2 and MaR1 each induced the efficient phosphorylation of the central mediator of inflammation, GSK3β, at Ser9—a potent inflammation inhibiting event. The activation status of SGK1, an anti-inflammatory kinase,
3
was similarly enhanced. Phosphorylation of SYK, a TLR ligand and inflammatory enhancer,
43
pERK1/2, pSAPK/JNK and/or p38 MAPK were unaffected (data not shown). However, RVD1, RVD2 and MaR1 all augmented the phosphorylation of CREB at Ser133 in TLR4-engaged innate cells, while the NF-κB (p65) signal was diminished. Importantly, resolvins alone did not influence the activation state of any of the inflammatory signaling molecules examined.
MaR1, RVD1 and RVD2 promote the phosphorylation of GSK3β and CREB but reduce p65 phosphorylation. (a) Western blots of whole-cell lysates (10 µg protein) from LPS-stimulated and unstimulated primary human monocytes. Blots were probed with Abs to phospho-p65 (S536), pSGK1 (S422), pAkt (S473), pGSK3β (S9) and pCREB (S133), as well as total GAPDH as a loading control. Densitometric quantification was performed by determination of the ratios of (b) pSGK, (c) pGSK3β, (d) pAKT, (e) pCREB and (f) pNFκB to total GAPDH signal using Image J software. MaR1 (1 μM), RVD1 (30 nM) or RVD2 (1 μM) concentrations were used. Representative blots are presented. *P < 0.05, **P < 0.01, ***P < 0.001 compared with LPS 60 min. #P < 0.05, ##P < 0.01, ###P < 0.0001 compared with LPS 15 min.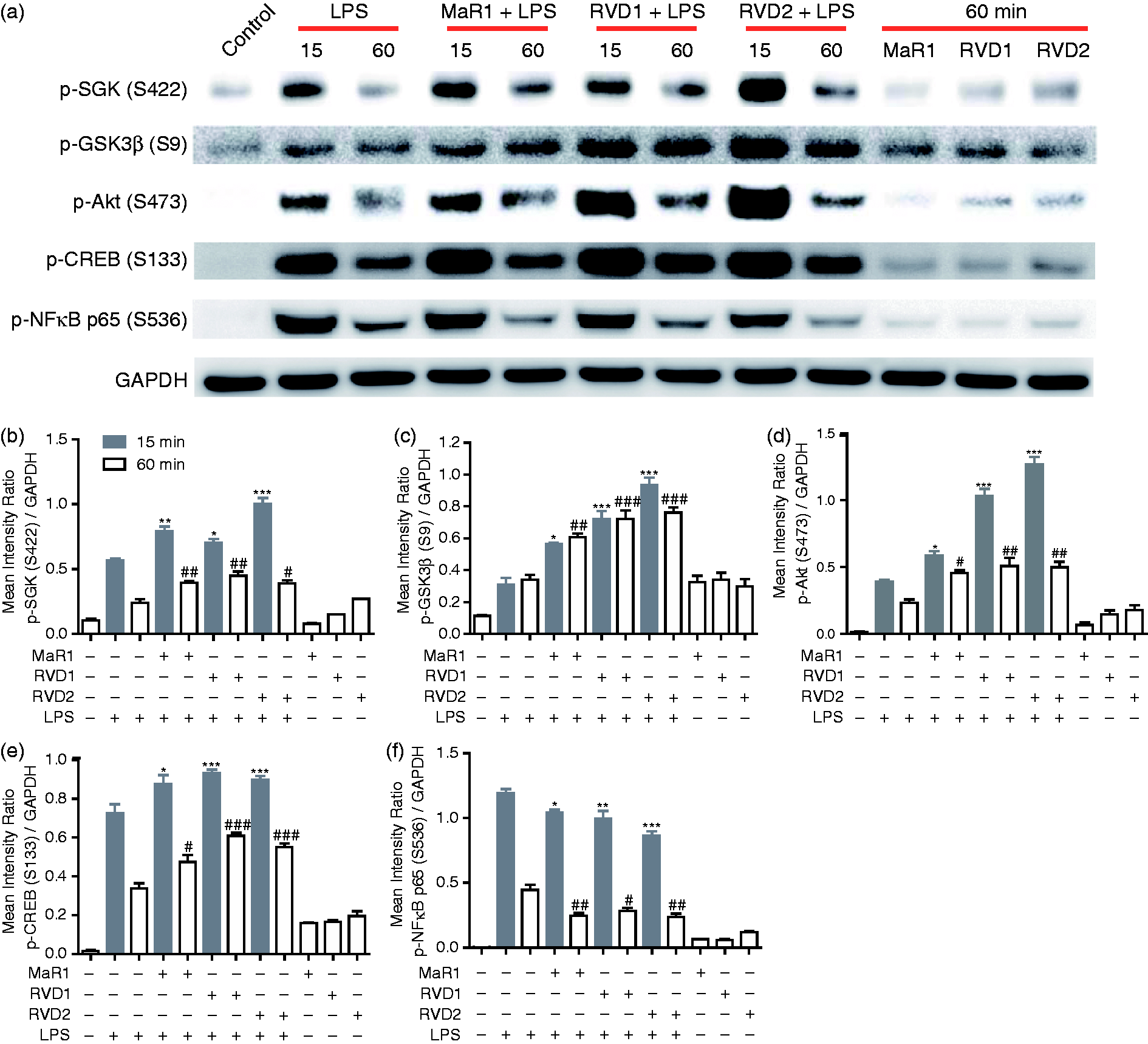
GSK3β(S9A) and GSK3β blockade alter the anti-inflammatory properties of MaR1
As all data suggested that resolvins function similarly (Figures 1–4), the mechanisms of resolvin-induced innate suppression were further elucidated using MaR1. The GSK3β inhibitor, SB216763, enhanced resolvin suppression of pro-inflammatory cytokine (IL-8; P < 0.05) production in the context of LPS concomitant with increased anti-inflammatory cytokine (IL-10; P < 0.001) secretion (Figure 5A, B). The augmentation by SB216763 suggests that inhibition of GSK3β by MaR1 is effective but incomplete. More importantly, however, MaR1 was unable to inhibit LPS-induced IL-8 production in monocytes transfected with pcDNA3-GSK3β(S9A) which expresses a GSK3β isoform that cannot be phosphorylatively inactivated (Figure 5C).
Inhibition of GSK3β or expression of GSK3βS9A alter the inflammatory response to LPS in MaR1-treated monocytes. (a) IL-10 and (b) IL-8 concentrations in 20 h cell-free supernatants were collected from primary human monocytes stimulated or not with LPS (1 µg/ml) in the presence or absence of MaR1 (1 μM) with and without GSK3β blockade by SB216763. (c) IL-8 release into 20-h cell-free supernatants of primary monocytes containing a plasmid expressing constitutively active GSK3βS9A (or empty vector) and stimulated with or without LPS (1 µg/ml) in the presence or absence of MaR1 (1 μM). Data represent the arithmetic mean ± SD of three biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001.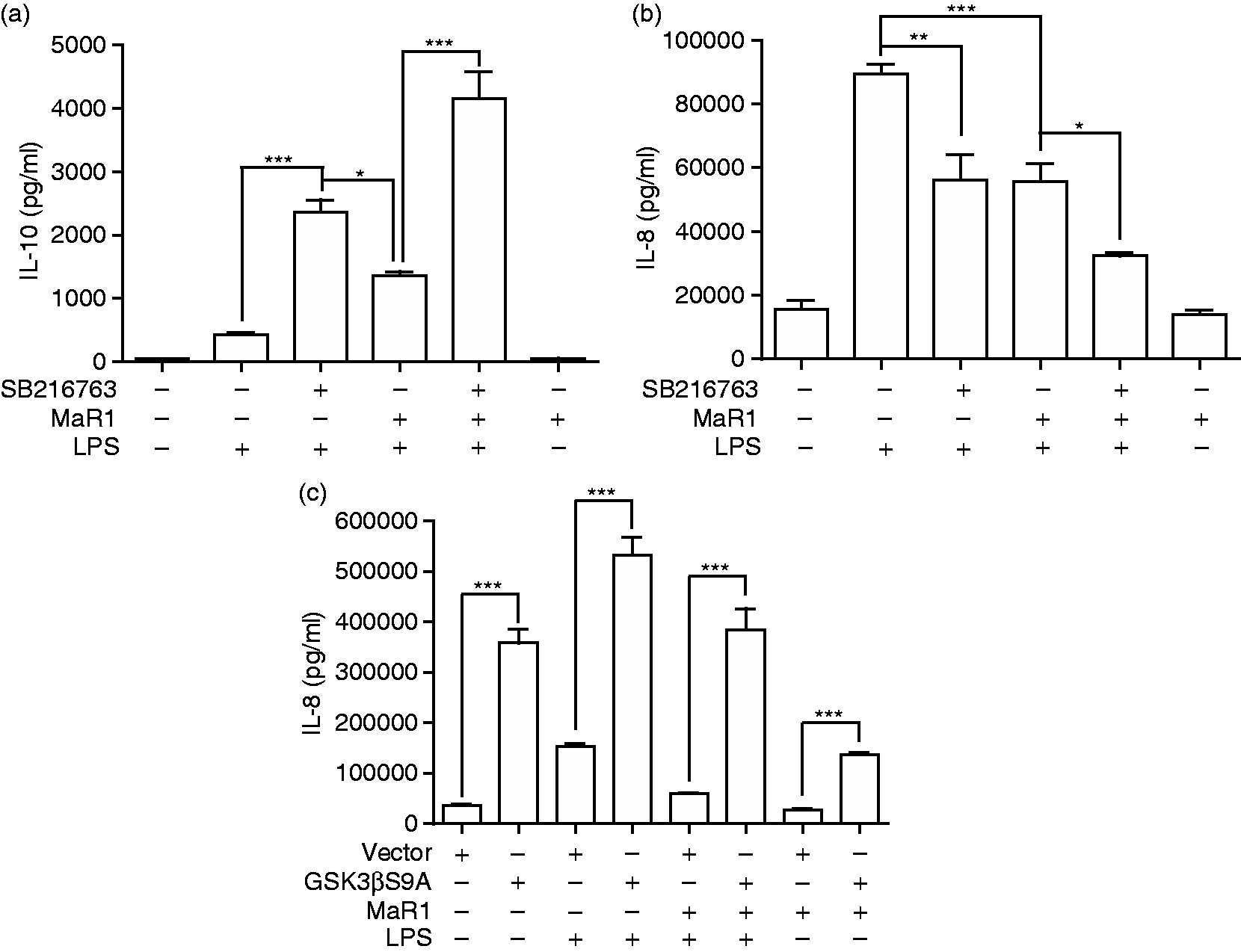
CREB silencing promotes a pro-inflammatory axis
As presented in Figure 6, siRNA-mediated creb silencing confirmed the functional role of CREB in resolvin-mediated augmentation of anti-inflammatory IL-10 production in TLR4-engaged monocytes. CREB levels were successfully knocked down by transfecting innate cells with creb-specific siRNA (Figure 6A, B). Such creb silencing led to the enhancement of pro-inflammatory cytokine (IL-8; P < 0.05) release concomitant with suppression of anti-inflammatory cytokine (IL-10, P < 0.001) secretion in MaR1- and LPS-exposed monocytes (Figure 6C, D).
CREB silencing abrogates MaR1-mediated IL-10 enhancement and Il-8 suppression in response to LPS. Successful knockdown of CREB by siRNA was confirmed by (a) Western blot and (b) densitometry. (c) IL-10 and (d) IL-8 concentrations in 20 h cell-free supernatants were collected from primary human monocytes stimulated with or without LPS (1 µg/ml) in the presence or absence of MaR1 (1 μM) with and without creb silencing. (a) and (b) are representative data. (c) and (d) are presented as the arithmetic mean ± SD of three biological replicates. *P < 0.05, ***P < 0.001.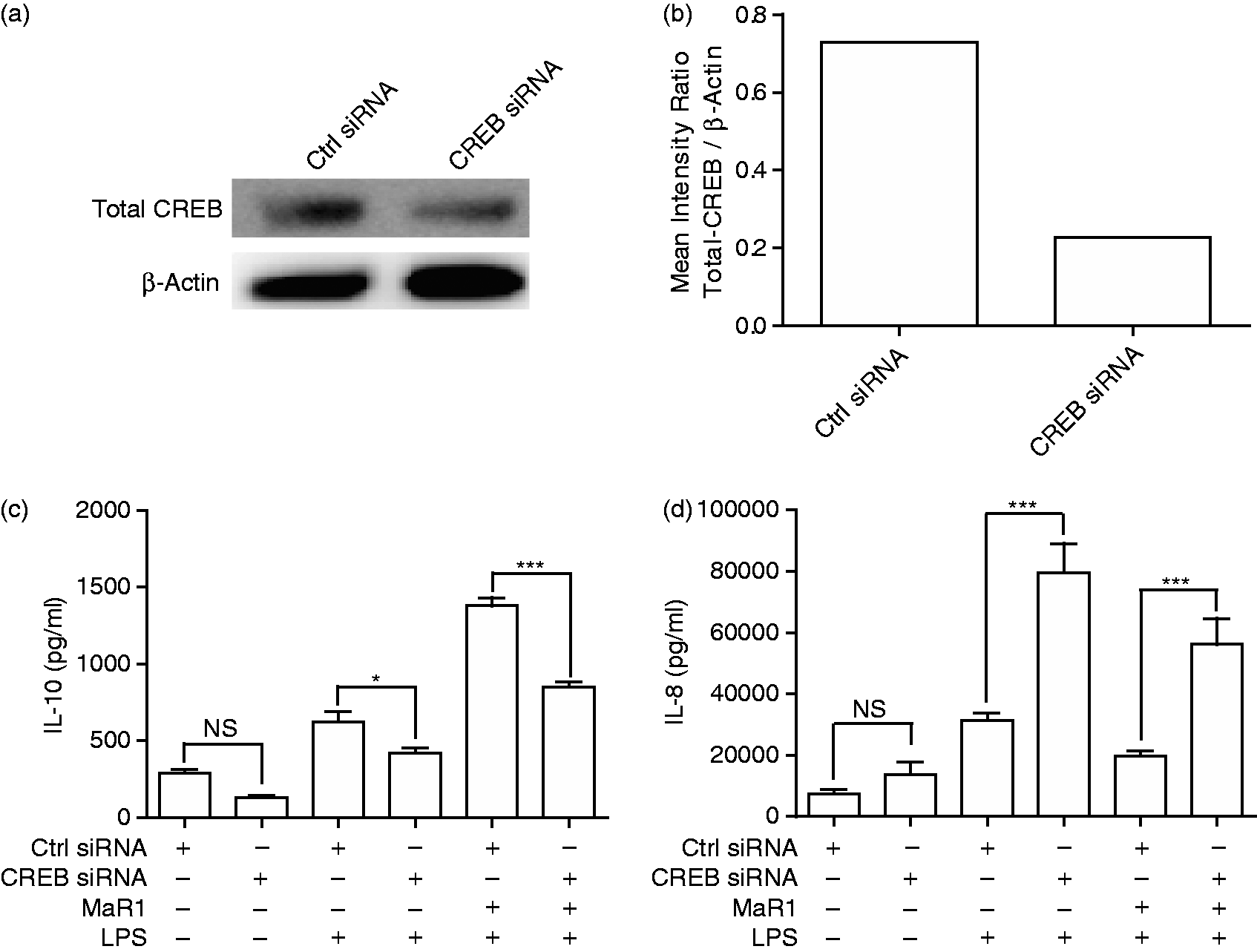
Discussion
Several key studies have established a critical role for endogenous and exogenous docosahexaenoic acid-derived lipid mediators, which appear to be share a degree of receptor commonality,44–46 in the resolution of inflammation.7,8,47,48 Interestingly, Hsiao et al. have suggested that RVD1 may increase the concentration of the anti-inflammatory cytokine, IL-10, in the lungs of smoke-exposed mice. 49 We show that RVD1, RVD2 and MaR1 are each efficacious inhibitors of multiple pro-inflammatory cytokines (IL-1β, IL-8, TNF) produced by monocytes in response to stimulation with the classic TLR-4 agonist, LPS. We also show, for the first time, that all three resolvins suppress the innate/adaptive bridging, T-cell-stimulating and angiogenic cytokine, IL-12 p40, while concomitantly increasing the expression of IL-10, the classic anti-inflammatory cytokine.
In order to harness and refine the anti-inflammatory therapeutic potential of resolvins, it is first necessary to understand their mechanisms of action. To this end, Nordgren et al. 28 have reported that while MaR1 pretreatment did not alter the activation of NF-κB, AP-1 or SP-1 in pollution particle-stimulated bronchial epithelial cells it did alter serum response element signaling. However, pollution particles may activate the innate response in a different manner than microbe-associated molecular patterns, and most studies have found an inhibitory effect of resolvins on the key pro-inflammatory transcriptional activator, NF-κB.32–35 Gilbert et al. 39 have reported that RVD1 promotes Akt activation and that the efficacy of RVD1 to suppress myocardial ischemia/reperfusion injury is abrogated by the PI3K inhibitor, LY-294002. Of the resolving mediators, RVE1 and LXA4 are among the better characterized, from a signaling perspective. Ohira et al. 41 have also reported that RVE1 can induce Akt–mTOR pathway activation, even in the absence of a primary pro-inflammatory mediator, in Chinese hamster ovary cells expressing the RVD1 receptor, Chem 23. 41 Prieto et al. 42 have established that LXA4 can activate the PI3K/Akt axis in macrophages. Wang et al. 16 have shown that the PI3K inhibitor (LY294002) blocks RVD1-induced expression of epithelial sodium channel α (ENaCα) in vitro and reduced the efficacy of RVD1 in suppressing pulmonary inflammation in a LPS-induced acute lung injury model. Other reports have also suggested RVD1 action may be mediated by PI3K. 40 PI3K is an activator of Akt, while Akt is an established phosphorylative inactivator of the central mediator of inflammation, GSK3β.1,2,38 Furthermore, the ratios of NF-κB and CREB help direct the direction and intensity of the inflammatory response, with activity of the IL-10 gene being driven, in part, by interactions of CREB with the IL-10 cAMP-responsive element. 38
We have augmented the PI3k–Akt-related literature by establishing that RVD1, RVD2 and MaR1 each enhance the phosphorylation of GSK3β (inactivation) and CREB (activation), while reducing the amount of phosphorylated p65 relative to pCREB or GADPH. Interestingly, the activation state of SGK1, which we have recently established to be a potent anti-inflammatory mediator in the context of TLR-engagement, 3 was also enhanced by MaR1, RVD1 and RVD2. However, the phosphorylation state of SYK, a TLR ligand and inflammatory enhancer, 43 and several MAPK-related molecules was not influenced by resolvins. All Abs employed were established and commonly employed commercial preparations. However, cross-reactivity with non-target proteins remains a possibility. To this end, it was important to verify key immunoblot-derived data by a combination of pharmaceutical inhibition, gene silencing and gain- or loss-of-function approaches. Plasmid-derived GSK3βS9A, which cannot be phosphorylatively inactivated, abrogated the anti-inflammatory capability of MaR1. To further confirm the importance of CREB in resolvin-enhanced IL-10 response, siRNA-mediated creb silencing resulted in inflammatory polarization in the opposite direction to the GSK3β inhibitor SB216763, i.e., creb gene silencing enhanced pro-inflammatory cytokine (IL-8) release while suppressing MaR1 augmentation of anti-inflammatory IL-10 production in TLR4-engaged monocytes.
We have examined innate suppression in the context of a professional cytokine-producing cell type (monocytes) engaged by a highly potent TLR agonist (E. coli LPS), an in vitro system that has required the utilization of relatively high doses of resolving, with IC50 values generally in the μM range. By comparison, inhibitory concentrations of resolving agonists used to suppress capsaicin (100 nM)-induced transient receptor potential vanilloid 1 currents in neurons (IC50 ≈ 0.5 nM) 50 or to reduce the TNF-induced IL-1β transcript signal in glioma cells (IC50 ≈ 50 pM) 51 are much lower. Others, however, have employed micromolar concentrations when determining the efficacy of pro-resolvins in the inhibition of cytokine production following direct engagement of TLRs in dedicated innate cells.29,30 Nevertheless, it remains to be seen if the same phenomena demonstrated in vitro herein occur in vivo.
There are many potential mechanisms of action of pro-resolving mediators, as recently reviewed.46,51–54 We have focused on the TLR4 (LPS)-GSK3β axis and established that resolvins appear to induce the GSK3β anti-inflammatory axis leading to enhancement of the active form of the IL-10 transcriptional enhancer, CREB. Further elucidation of common and divergent mechanisms of action of such resolvins should facilitate the translation of RVD1, RVD2 and MaR1 for the amelioration of infection-driven inflammation and, perhaps, other conditions associated with dysregulation of the innate immune response. Indeed, it may even be possible to manipulate resolvin-related signaling events in order to up- or down-regulate the direction and intensity of the innate response, depending on the clinical necessity.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by NIDCR grants DE017680 (DAS), DE023633 (HW), DE025409 (SMU) and DE017921, DE011111 (RJL).