
Abstract
Cardiovascular diseases are chronic inflammatory diseases that affect a large segment of society. Coronary heart disease (CHD), the most common cardiovascular disease, progresses over several years and affects millions of people worldwide. Chronic infections may contribute to the systemic inflammation and enhance the risk for CHD. Periodontitis is one of the most common chronic infections that affects up to 50% of the adult population. Under inflammatory conditions the activation of endogenous degradation pathways mediated by immune responses leads to the release of destructive cellular molecules from both resident and immigrant cells. Matrix metalloproteinases (MMPs) and their regulators can activate each other and play an important role in immune response via degrading extracellular matrix components and modulating cytokines and chemokines. The action of MMPs is required for immigrant cell recruitment at the site of inflammation. Stimulated neutrophils represent the major pathogen-fighting immune cells that upregulate expression of several proteinases and oxidative enzymes, which can degrade extracellular matrix components (e.g. MMP-8, MMP-9 and neutrophil elastase). The activity of MMPs is regulated by endogenous inhibitors and/or candidate MMPs (e.g. MMP-7). The balance between MMPs and their inhibitors is thought to mirror the proteolytic burden. Thus, neutrophil-derived biomarkers, including myeloperoxidase, may activate proteolytic destructive cascades that are involved in subsequent immune-pathological events associated with both periodontitis and CHD. Here, we review the existing studies on the contribution of MMPs and their regulators to the infection-related pathology. Also, we discuss the possible proteolytic involvement and role of neutrophil-derived enzymes as an etiological link between chronic periodontitis and CHD.
Keywords
Introduction
Chronic periodontitis
Chronic periodontitis (CP), the most prevalent form of periodontitis, is defined as an inflammatory disease of the tooth-supporting structures. CP is caused by a complex interplay between host defense and biofilm dysbiosis indicated by growth of specific pathogens or complexes of pathogens colonizing the subgingival area. To challenge the microbial biofilm and its virulence factors (LPS, enzymes and toxins), an immune-inflammatory response develops. Resident tissue cells induce and produce pro-inflammatory mediators, which enhance the recruitment of inflammatory cells (primarily neutrophils) at the site of inflammation. Once neutrophils reach the inflamed sites, they aggregate, form a wall separating the epithelium from the bacterial biofilm, degranulate large quantities of tissue destructive enzymes [e.g. matrix metalloproteinase (MMP)-8, MMP-9 and neutrophil elastase (NE)] and generate ROS.1–3 These cascades result in collagen loss and further progressive breakdown of soft and hard tissues of the periodontium, leading to pocket formation and/or recession. If not treated, CP eventually leads to tooth loss.4–6
When periodontitis progresses the predominance of Gram-positive bacterial species in the biofilm changes majorly into Gram-negative species.7,8 According to culture and DNA hybridization techniques, Porphyromonas gingivalis, Prevotella intermedia, Treponema denticola, Tannerella forsythia and Aggregatibacter actinomycetemcomitans are considered to be etiologically linked with periodontal diseases (PDs) or frequently found in pathological periodontal conditions.7,8 Culture-independent studies have expanded the range of disease-associated organisms, 9 and around 1000 bacterial species have been found in the oral cavity. 10 Periodontal bacteria or their products, for example LPS, may enter the bloodstream through inflamed periodontal tissues, especially after dental treatment,11–13 gentle mastification or tooth brushing.12,14 Similarly as during the acute-phase response, in periodontitis patients and... during the acute-phase response in periodontitis patients and in a mouse model infected with A. actinomycetemcomitans,15–17 most of the endotoxin activity is found in the pro-atherogenic lipoprotein fraction. This alteration is considered to promote pro-atherogenic properties of the lipoproteins, including activation of macrophages and accumulation of cellular cholesterol. 18 Long-term or repeated episodes of bacteremia and endotoxemia are undoubtedly threats to general health in both healthy people and those with metabolic disorders.19–24
Coronary heart disease
Coronary heart disease (CHD) progresses over several years and affects millions of people worldwide. The disease may lead to acute coronary syndrome (ACS) [unstable angina pectoris and myocardial infarction (MI)] which is considered as the major cause for mortality in patients with cardiovascular diseases (CVD). 25 Atherosclerosis is the underlying cause for CHD and represents a multifactorial degenerative disease of large- and medium-sized arteries. It leads to lipid-rich plaque formation, artery wall thickening and atheroma development. The incidence of atherosclerosis cannot be fully explained by classical risk factors. The hypothesis of infection as a potential cause of atherosclerosis has gained favour and is supported by a large body of epidemiological evidence.25–28 Inflammation is thought to contribute to the progression of atherosclerotic lesions and may also have a fundamental role in thrombosis and adverse acute outcomes causing death. 29
Association of CP and CHD
Since Mattila et al. addressed an association between oral infections and CHD in 1989, 30 numerous epidemiologic studies have revealed a link between CP and CVD.31–39 Although the findings were modest and no causal association could be found, these studies suggested an independent consistent association that cannot be attributed to common risk factors. 37
CP, like other life-long infectious diseases, may affect initiation, development and progression of CHD either directly by bacterial vascular invasion or indirectly through systemic inflammation or antigen cross-reactivity (Figure 1).40–44 Most importantly, successful treatment of periodontitis has positive effects on CVD-associated risk factors.45–47
Schematic representation of principal interactions of neutrophil-derived proteases and the anti-protease shield, addressing a possible mechanistic link between CP and acute manifestations of CVD.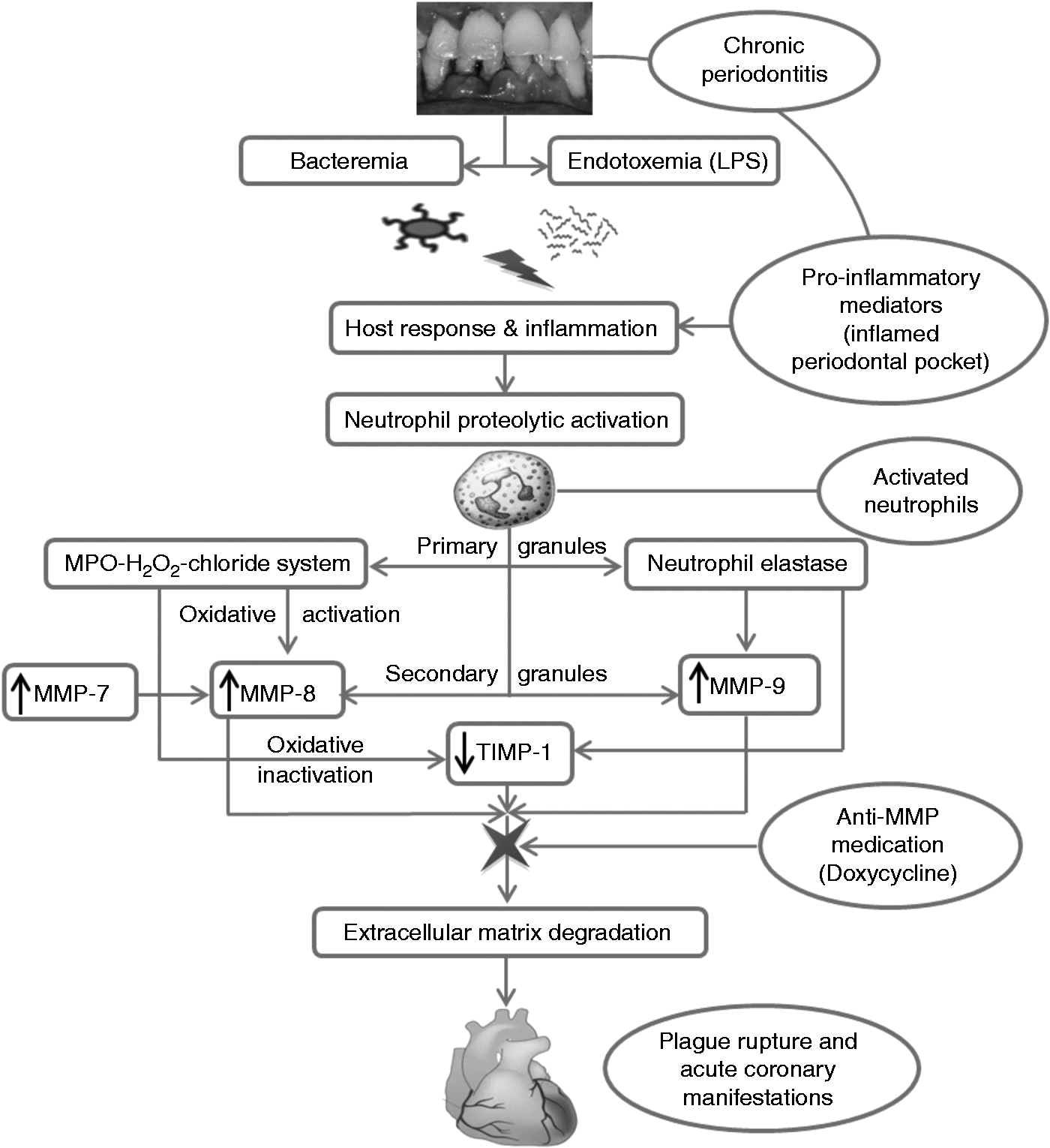
Bacteria may access the circulation during daily routine, oral hygiene procedures and during periodontal therapy.22,48,49 The epithelial ulceration at the periodontal pocket confers direct access of virulent Gram-negative organisms (e.g. P. gingivalis and A. actinomycetemcomitans) to the blood stream that causes recurrent and transient bacteremia, as well as low-grade systemic inflammation.12,13,50 Trafficking of phagocytes represents another direct route that periodontal pathogens may circulate in the blood stream, invade endothelial cells and possibly promote atherosclerosis-related vascular inflammation. 51 Systemic challenge to periodontal pathogens and their soluble components induces a major vascular response, which may alter the endothelial integrity; this represents the earliest change in the vascular wall, followed by leukocyte aggregation, cholesterol deposition, atheroma formation and progression, and plaque rupture in further consequence. These hypotheses are confirmed by the detection of multiple periodontal pathogens, as well as their identification at the DNA level, in human atherosclerotic plaques.29,52–54
Host inflammatory response and molecular mimicry represent another indirect mechanism linking CP and CHD. Periodontal pathogens and virulence factors are capable of inducing systemic inflammation, which, in turn, affects all stages of the atherosclerotic process. Locally secreted pro-inflammatory cytokines such as TNF-α, IL-1 and IL-6 enter circulation, trigger the release of acute-phase reactants (e.g. C-reactive protein) and promote cell activation. This leads to production of adhesion molecules, activation of TLRs and the release of MMPs (e.g. MMP-9) and their regulators [e.g. tissue inhibitors of metalloproteinase-1 (TIMP-1)] and NE, respectively—processes accelerating the development of the atherosclerotic process in the vessel wall.41,43,55–57 Cross-reactive auto-antibodies against common antigens of periodontopathogens and the host (e.g. heat shock proteins) generated by molecular mimicry may disturb the immune reaction and contribute to the pathogenesis of PD and CHD, probably via similar activation processes and cascades.58,59
Appropriate periodontal treatment has been reported to be effective in reducing and improving markers associated with CVD, for example CRP, IL-6, TNF-α, cholesterol levels and endothelial dysfunction.45–47 Importantly, periodontal treatment may have a beneficial effect also on the function and properties of all lipoprotein classes.13,18,60 Thus, these reports point towards a causal association between CP and CHD.
A large body of evidence reporting possible pathogenic pathways that may link PD with CHD has been published. The present review will primarily cover the proteolytic role of MMPs and their regulators, as well as the role of myeloperoxidase (MPO), a neutrophil-derived enzyme that gets upregulated during inflammatory diseases, 61 including atherosclerosis, glomerulosclerosis, glomerulonephritis and PD (see below).62–67
MMPs and their regulators in periodontitis and CHD
The MMP family consists of at least 23 genetically distinct but structurally related zinc- and calcium-dependent endopeptidases, which cooperatively participate in a protease cascade to remodel almost all extracellular matrix (ECM) and basement membrane (BM) constituents. MMPs can process a number of soluble proteins such as cytokines, chemokines and growth factors, and activate individual MMPs, thus generating cascade-type MMP-dependent immune responses (Figures 1 and 2).68–71 Alternatively, pro-MMPs in cascade can also be activated by microbial proteases, serine proteinases and ROS (Figures 1 and 2). The ability of MMPs to cleave ECM components and to regulate the activity of non-ECM bioactive molecules confers their crucial roles in various physiological and pathological processes such as tissue development, immune responses, remodeling, and in inflammatory and vascular diseases. MMPs are often categorized according to their modular domain structure: (i) collagenases (MMP-1, MMP-8 and MMP-13); (ii) gelatinases (MMP-2 and MMP-9); (iii) stromelysins (MMP-3, MMP-10 and MMP-11); (iv) matrilysins (MMP-7 and MMP-26); (v) membrane-type MMPs (MMP-14, MMP-15-17, MMP-24 and MMP-25); and (vi) others (MMP-12, MMP-19 to MMP-21, MMP-23, MMP-27 and MMP-28).68,72
Proteolytic and oxidative activation cascades of MMP-8 and MMP-9 associated with periodontitis and CVD. This activation cascade can be potentiated by microbial proteases, serine proteases, other MMPs and MPO but will be inhibited by TIMP-1.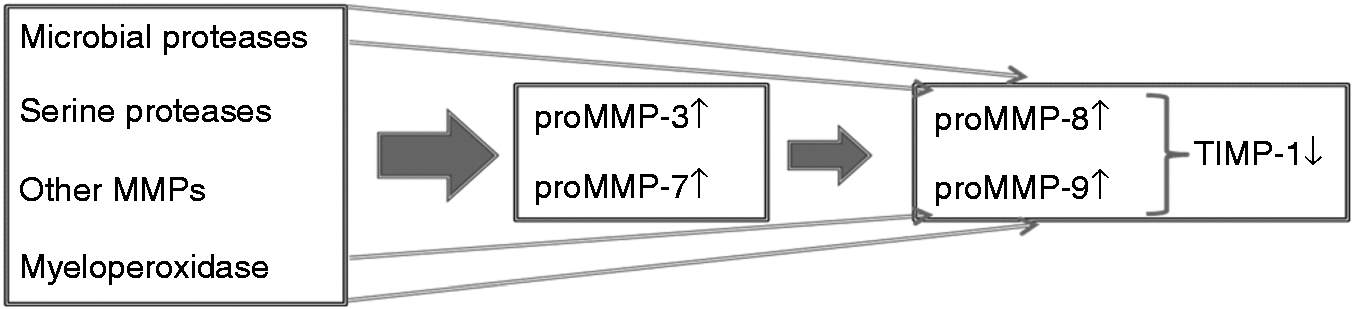
MMP-8 (collagenase-2)
Upon maturation in the bone marrow, neutrophils (also called polymorphonuclear leukocytes) synthesize and store MMP-8 (also called neutrophil collagenase) in secondary granules as a latent enzyme. In response to various extracellular stimuli neutrophils degranulate and release mature MMP-8. Owing to its collagenolytic properties, MMP-8 is contributing to leukocyte recruitment at the site of inflammation. In addition to neutrophils (the main source of MMP-8), a variety of cells of the non-polymorphonuclear leukocyte lineage, such as monocytes/macrophages, plasma cells, endothelial cells, fibroblasts and epithelial cells, can express MMP-8 during inflammatory processes.2,68–70,72–76 MMP-8 can initiate the cleavage of collagen type I–III; most importantly, MMP-8 has a high affinity for type I collagen. Other MMPs, like gelatinases can further cleave the proteolytic products of collagens after the initial cleavage by collagenases.69,70,77–80
Collagen type I is the preferred substrate for MMP-8 that is secreted by different inflamed cells in the atherosclerotic plaque. Type I is the most abundant collagen and the major load-bearing molecule in the atherosclerotic fibrous cap; it is obvious that MMP-8 plays a crucial role in atherosclerosis. 81 Furthermore, MMP-8 degrades apolipoproteins A–I (apoA–I; the major apolipoprotein of high-density lipoproteins) thereby decreasing the anti-atherogenic function of lipoproteins of the high-density range during reverse cholesterol transport. 82 MMP-8 has been proposed to act as a suitable marker for cardiovascular outcomes.83–86 An increased expression of MMP-8 was found in activated inflammatory cells covering the shoulder region of the atherosclerotic plaque.81,87,88 MMP-8 has been found to be released excessively in patients with CVD characterized by plaque progression, 89 to be significantly elevated in ruptured infarct tissue in patients with MI, 86 to potentially reflect coronary plaque instability in patients with unstable angina pectoris, 90 and to be linked with the severity of coronary artery disease and ACS.91,92 In addition, high serum MMP-8 levels are associated with ACS, 93 particularly in patients with acute MI, CVD and cardiac arrest.86,94–96
MMP-8 is the predominant MMP associated with periodontitis.69,70 Periodontal pathogens and their virulence factors can activate resident cells to generate inflammatory mediators,97–99 by which the latent MMP-8 can be proteolytically and oxidatively activated. 84 Active MMP-8 can degrade type I collagen, the major component of the ECM in the periodontal tissues, leading to undesired destructive lesions. Saliva and gingival crevicular fluids (GCF) are easily and noninvasively collectable diagnostic biological specimens that are useful for the detection of early periodontitis. Monitoring of candidate fluid biomarkers for both oral and systemic conditions is a necessary tool to complement clinical examinations in epidemiological studies. Nonetheless, it should be noted that smoking, variations in the salivary flow rates and other factors may affect the usefulness of oral fluid marker analyses, and thus the levels should be considered with caution when interpreting results. 100 Several studies have shown that MMP-8 levels in oral fluids correlate with the severity of periodontal inflammation, being elevated especially in severe periodontitis.85,101–109 MMP-8 levels decrease in response to different periodontal treatments such as scaling, root planning or application of the collagenase inhibitor doxycycline.103,110 Kivelä-Rajamäki et al. even reported that high MMP-8 levels have been identified in an active form in diseased peri-implant sulcular fluids. 111 To conclude, MMP-8 plays an important role not only in periodontal tissue destruction, but also in periodontal homeostasis and defense.112,113 However, future studies are required to address whether salivary or mouth rinse analysis of MMP-8, MMP-9, MPO and their regulators can be utilized as potential diagnostic tools in systemic diseases such as CVD.56,57
MMP-9 (gelatinase B)
Similarly as reported for MMP-8, MMP-9 is mainly synthesized by neutrophils and stored in their intracellular secondary granules as a latent enzyme. 114 Upon bacterial challenge activated leukocytes migrate to the site of inflammation and secrete MMP-9 as a latent form, which is activated locally by trypsin, α-chymotrypsin, cathepsin G, plasmin and other MMPs (e.g. MMP-3, MMP-7 and MMP-10) by removal of the pro-peptide (Figure 2).115–117 In addition to neutrophils, MMP-9 is also secreted by macrophages, smooth muscle cells (SMCs), epithelial and endothelial cells.115,118–122 In order to facilitate leukocyte migration, expression and activation of MMP-9 is increased during inflammatory processes, including periodontitis.69,70,123–125 In addition to gelatins, MMP-9 cleaves also ECM and BM components such as collagen type IV/V, aggrecan, elastin and other substances, for example IL-1β.122,126–130 Expression of MMP-9 can be induced by MMP-7, as well as different cytokines, including IFN-γ, IL-1, IL-2 and TNF-α, respectively.
MMP-9 plays a subtle role in the progression of CVD. Expression of MMP-9 has been described in macrophages, SMCs and endothelial cells derived from atherosclerotic plaque material, particularly at the shoulder region. Furthermore, MMP-9 cleaves type IV collagen and denatured collagen, and may contribute to plaque formation and destabilization via facilitation of medial SMC migration to the intima. Thus, it contributes to degradation of the thin collagen cap that covers cholesterol-rich plaques lining the coronary arteries, which leads to plaque rupture, thrombosis and acute MI.88,96,131–134 The active form of MMP-9 is elevated in clinically defined unstable carotid plaque, and MMP-9 levels are significantly higher in ruptured infarct tissue in patients with fatal MI.84,86,135 Furthermore, elevated serum or plasma levels of MMP-9 have been reported in patients with cardiac arrest, unstable angina and acute MI, or patients with a history of MI.96,136–138
MMP-9 is considered as one of the major MMPs expressed in periodontitis-affected gingiva. This protease has been found to be associated with severe periodontitis but its levels decrease after successful periodontal intervention.69,70,125,139 Elevated serum levels of MMP-9 have also been reported in patients with periodontitis, but decreased significantly after 3 months of non-surgical periodontal intervention.57,106 One cross-sectional study showed that GCF levels of MMP-8 and MMP-9 correlate with disease activity in patients with PD. 105
MMP-7 (matrilysin-1)
Owing to the absence of the hemopexin-like domain that is common to all other MMPs, MMP-7, the smallest (28 kDa) of the known MMP family members, is less susceptible to inhibition by TIMPs.140–143 Epithelial MMP-7 is secreted in its latent form and can be effectively activated by plasmin and MMP-3 through proteolytic removal of the pro-domain.115,144 MMP-7 is secreted by various cells, including epithelial cells and macrophages, but not by neutrophils, and can degrade elastin, laminin, collagen type IV and IX and fibronectin. MMP-7 cannot cleave interstitial collagen, but it can activate latent forms of other MMPs (pro-MMP-1, pro-MMP-2, pro-MMP-8 and pro-MMP-9) (Figure 2), and thus potentiates proteolytic-cascades. MMP-7 plays a key role in both epithelial repair and defense, 140 and it may have a specific role in the intraepithelial cell migration process during renewal of the epithelium when expressed by junctional epithelium.111,140,145–147 In addition, MMP-7 may express antimicrobial defense properties in response to bacterial insult by converting antimicrobial pro-defensin peptides into their active forms. 140
MMP-7 is also expressed by lipid-laden macrophages in atherosclerotic lesions, and serum MMP-7 levels are elevated in cardiac arrest patients compared with healthy controls. 96 Increased MMP-7 expression was found in macrophages and SMCs covering the shoulder regions of the atherosclerotic plaque. 148 Plasma MMP-7 concentrations are elevated in patients with (un)stable coronary artery disease and CVD.94,149 Furthermore, MMP-7 may contribute to collagenolysis preceding the atherosclerotic plaque rupture by cleaving pro-MMP-8 into active MMP-8. 150
MMP-7 is induced by microbial products such as LPS. 140 Furthermore, MMP-7 was found to be expressed by periodontitis-affected human gingival sulcular epithelium in vivo and in peri-implant sulcular fluid.111,151 MMP-7 is released in gingival tissues of patients with periodontitis and is elevated in CP.145,151
TIMPs
TIMPs (TIMP-1–4) are natural MMP inhibitors. Similar to MMPs, TIMPs are expressed at low levels in normal tissues. However, TIMP expression rises during tissue remodeling under both physiological and pathological conditions.152,153 Although TIMPs can inhibit all individual MMP proteins, the inhibitory effect varies among the different MMP species. Owing to the lack of the hemopexin domain various MMPs are less tightly bound to TIMPs, and therefore are poorly inhibited. 154 The activity of MMP is inhibited via non-covalent binding of the N-terminal portion of TIMP to the C-terminal portion of MMP (Figures 1 and 2).68,71,146,153,154 The imbalance between MMPs and their TIMP inhibitors leads to an excessive and undesirable tissue destruction at the site of inflammation.69,70
Various vascular tissue cells (e.g. endothelial cells, monocytes/macrophages and SMCs) can express TIMP-1. TIMP-1 expression and its release by neutrophils are very low or hardly detectable.155,156 Through inhibiting MMPs, TIMP-1 blocks SMC migration and thus plays a beneficial role against progression of atherogenesis. 115 High serum TIMP-1 levels have been correlated with plaque instability and tissue damage and various forms of CVD (e.g. MI and cardiac mortality),157,158 and may predict fatal future outcomes.93,94 The MMP/TIMP ratio may be considered an important parameter affecting atherogenesis. 137 Furthermore, an increased MMP-8/TIMP-1 ratio was found to be strongly associated with ACS and CVD,93,137 and thus may be considered as a predictor for coronary risk. 94
TIMP-1 expression is found in several periodontitis-affected gingival tissue cells such as endothelial cells, monocytes/macrophages, keratinocytes and fibroblasts. 159 In oral samples, the balance between MMPs and TIMPs is regarded to mirror the proteolytic burden.69,70 Low TIMP-1 levels have been demonstrated in patients with periodontitis compared with controls, and after periodontal treatment these levels appear to rise. Furthermore, the MMP-8/TIMP-1 ratio in salivary and GCFs can be used to discriminate patients with periodontitis from controls.160–162
NE
NE (also termed polymorphonuclear leukocyte elastase) is a serine protease. Activated neutrophils express and store NE in their primary granules in order to combat bacterial insult, but the enzyme may also contribute to undesired tissue degradation.163,164 NE can degrade elastin, collagen type I–IV, laminins, fibronectin and proteoglycans.79,163,164 Moreover, NE is able to degrade ECM by accelerating MMP cascades. NE is also able to activate pro-MMPs, such as pro-MMP-9 and pro-MMP-3, and to inactivate TIMPs, a process that essentially modulates the MMP/TIMP ratio.165,166 NE can modulate the activity of various cytokines and favor thrombus formation.116,167 As macrophages present in human lesion material express NE, 168 an enzyme that has been suggested to be associated with an increased risk of CVD and plaque instability.94,169,170
Furthermore, NE can degrade non-collagenous protein-covered collagen fibrils in the early destructive stages of PDs. 171 High serum levels of NE were reported in untreated periodontitis than in periodontally healthy controls. 172 Most importantly, serum NE levels were significantly decreased in response to non-surgical periodontal intervention. 173
MPO
Another circulating biomarker for CVD/CHD used to predict clinical risk and prognosis of affected patients is MPO.174–179 The predominant in vivo sources for MPO released during the oxidative burst are neutrophils and monocytes, in which MPO makes up to 5% and 1% in the total cell protein content, respectively. Furthermore, a subpopulation of macrophages expressing MPO is considered to play a particular role in atheroma complication and ACS.180,181 MPO generates the potent oxidant hypochlorous acid (HOCl) from H2O2 and chloride ions. Besides its antimicrobial activity, MPO contributes to degradation of connective tissues by impairing the crucial balance between proteases and anti-proteases. MPO is also present in the ECM in human lesion material; 64 the enzyme co-localizes with HOCl-modified epitopes/proteins at the endothelial layer and macrophages,182,183 and most importantly also with MMP-7. 184 As shown by Weiss and coworkers, with pro-MMP-8 and pro-MMP-9,185,186 and later by Fu et al., 184 with pro-MMP-7, HOCl may rapidly activate these zymogens, a process apparently depending on the oxidant:enzyme molar ratio. 187 This suggests that HOCl formed by the MPO-H2O2-halide system of activated phagocytes provides an oxidative mechanism for activating latent MMPs in vascular diseases; a pathway that may play a critical role in the rupture of atherosclerotic lesions,184,188 regulation of neutrophil recruitment and inactivation of TIMPs (Figures 1 and 2). Indeed, in vitro studies have shown that HOCl added as reagent or generated by the MPO-H2O2-chloride system inactivates TIMP-1 by oxidizing the N-terminal cysteine residue of the enzyme,189,190 a mechanism reported to occur in vivo under inflammatory conditions. 190 These data support the notion that an imbalance between the proteolytic activity of MMPs and the inhibitory activity of TIMPs is implicated in many pathological conditions. 190
A large body of data reported increased neutrophil infiltration during CP, reflecting a heightened inflammatory state. As a consequence, MPO mass and/or activity is upregulated in rapidly progressing CP, GCFs, saliva and human dental pulp tissues in order to combat pathogenic microbes.105,191–204 Miyasaki and Nemirovsky reported that among dental and periodontal bacteria tested, 204 A. actinomycetemcomitans has the highest capacity to promote the release of neutrophil-derived MPO. Owing to its ability to form HOCl, MPO is involved in the destruction of periodontal components and can destroy the ECM by directly activating latent MMP-8 and MMP-9, and by enhancing MMP activity via inactivation of TIMP-1. 101 Leppilahti et al. reported that levels of MPO and MMPs (MMP-8, MMP-13 and MMP-14) show highest diagnostic accuracy, 196 while only MPO and MMP-8 were significantly higher in periodontitis compared with gingivitis. Furthermore, salivary concentrations of MPO and NE, and the ratio of MMP-8/TIMP-1, were higher in generalized CP and aggressive periodontitis than in healthy controls. 202 The high prognostic value of MPO and MMPs, as well as TIMPs, in periodontitis is supported by a marked decrease even after non-surgical therapy.69,101,105,201 Basically, MPO levels in saliva are increased in patients with peri-implant disease with and without implants.205,206 In peri-implant sulcus fluids, levels of MPO rose with the increase of pocket probing depth and increasing gingival inflammation but decreased significantly after non-surgical therapy.207,208
Neutrophils, the first cellular responders to invading microbes, exert most of their antimicrobial activity in phagosomes, specialized membrane-bound intracellular compartments formed by ingestion of microorganisms. 209 Alternatively, stimulated neutrophils may combat microbes through the release of web-like filamentous structures of decondensed chromatin, so-called neutrophil extracellular traps. These traps are composed of DNA and histones, and harbor antimicrobial peptides and enzymes such as cathepsin G, NE and MPO. 210 Neutrophil extracellular traps are present in dental plaque samples, saliva, supragingival biofilms and gingival connective tissue,211,212 and thus play a significant role in the pathogenesis of periodontitis (for a review see Cooper et al. 211 ) probably by subsequent modulation of the immune response. 213
High levels of MPO–DNA complexes, as observed in severe coronary atherosclerosis, 214 are most likely due to periodontal bacteria (P. gingivalis, T. forsythia and P. intermedia) that may trigger neutrophil activation. This suggests activation of similar pathways as in atherosclerosis and periodontitis. 215 Treatment of periodontitis has further been reported to modulate the atherosclerotic profile by exerting a beneficial effect on endothelial cell function. 47 This must be seen in context: MPO-mediated endothelial dysfunction, apparently due to consumption of NO, 216 may be considered an important mechanistic link between inflammation and CVD. 217 Different MPO polymorphisms have been found to be associated with plasma MPO concentrations in patients with coronary artery disease. 178 Whether such a link may also exist with periodontitis needs further investigation.218–220
Neutrophil proteolytic activation cascades
Naruko et al. were the first to show neutrophil infiltration into culprit lesions in ACS. 221 Later, Ionita et al. reported that high neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. 222 Furthermore, a positive association between the number of neutrophils and plaque levels of MMP-8 and MMP-9 was found. 222 From these data it is obvious that neutrophils play a major role in mediating destabilization of the atherosclerotic plaque.221,222 Neutrophils are present in greater numbers in periodontal patients, and acute MI size is related to the extent of periodontitis. 223 Furthermore, neutrophils play a crucial role in the initiation and/or progression of both periodontitis and CHD, probably via proteolytic alteration in the local balance of ECM.1,2,76,224 Collagen structures of both periodontal ligament and atherosclerotic fibrous cap are almost the same. In the periodontium, mainly type I collagen and to a lesser extent type III collagen represent the main component of the ECM in the soft (gingiva and periodontal ligament) and hard (alveolar bone) periodontal tissues.225,226 Similarly, the atherosclerotic fibrous cap is rich in collagen type I and III.25,26,28,227 In order to approach the infected sites and eradicate the infectious bacterial burden, neutrophils release several proteases from their granules to degrade the collagen and gelatine moieties of the ECM and BM components, and to fulfill their antimicrobial function. 2 CP can result in enhanced production of neutrophil-derived proteases, both at local sites and also in the circulation. 2 The existence of periodontal bacteria in atheromatous plaque lesion may trigger neutrophil activation and recruitment at inflammatory sites. 215 Thus, proteolytic biomarkers released directly from neutrophils at the atheromatous plaque site or secreted in the circulation by neutrophils at sites of periodontal lesions may be considered a link between these inflammatory diseases (CP and CHD). Robust or prolonged neutrophilic antimicrobial activities may cause collateral uncontrolled destructive lesions.2,69,224 MMPs are the key players in this process by cleaving almost all ECM constituents and regulating the action of cytokines and chemokines.69–72,124,228
Doxycycline and other medications used for reducing MMPs or MPO and low-grade inflammation
Doxycycline, an approved adjunctive medication for the treatment of CP, is a synthetic MMP inhibitor and may be administered at three different pharmacological doses. It can be applied as either low- or submicrobial-dose doxycycline, 20–40 mg, or at a normal/regular dose (>100 mg). Owing to its anti-MMP properties, doxycycline may decrease the risk of coronary artery disease events. Doxycycline, at both low and regular doses, can downregulate several MMPs and other pro-inflammatory mediators, probably owing to its immunomodulatory and anti-proteolytic effects.42,162,229–231 In contrast, low or sub-antimicrobial adjunctive doxycycline medication does not exert antimicrobial properties, thus differing clearly from regular antimicrobial dosages. 232 Doxycycline can decrease MMP-7 and the MMP-8/TIMP-1 ratio, modulate NE, MPO, cytokines (IL-6, IL-8, TNF-α) and CRP, and most importantly increase TIMP-1 levels.42,133,139,162 Therefore, doxycycline seems to exert its potential as an adjunctive medication for multiple pathological conditions and chronic inflammatory diseases, including CP, CVD and ACS.85,133,139 In line with previous studies, our data have shown that MMP-7 and MMP-8, as well as the MMP-8/TIMP-1 ratio, were diminished with doxycycline treatment.42,161,229,233–235 Recently, at low doses, doxycycline has been suggested to decrease MMP-8 and MMP-9 levels in serum and oral fluids (such as GCF) and, consequently, beneficially modulate the MMP-8/TIMP-1 ratio.110,162,236 Frankwich et al. have demonstrated that regular-dose doxycycline medication decreased serum pro-inflammatory biomarkers and MMPs, resulting in improved insulin sensitivity eventually by protecting insulin receptor cleavage(s) by MMPs.237,238 Nevertheless, some adverse side effects (e.g. diarrhea, fungal infections and super infections) have been reported to occur when patients are treated with doxycycline at regular doses. 239
In rat models of experimental periodontitis, increased levels of MPO may be considered an inflammatory disease marker.240–242 Therapeutic effects on MPO levels by antioxidants such as alpha lipoic acid and vitamin C have been reported. 240 Both, carvedilol (an alpha/beta blocker) or azilsartan/olmersartan (angiotensin II receptor-AT1 blockers) reduced levels of MPO, MMP-2 and MMP-9.243–246 Treatment with synthetic parstatin (a 41-aa peptide, formed in vivo by proteolytic cleavage on activation of the protease activated receptor-1) significantly reduced MPO activity in gingivamucosal tissue. 242 Also, treatment of periodontitis with the tryptase inhibitor Nafamostat mesilate for 14 days decreased MPO activity in gingivomucosal tissue. 247
The combination of alendronate (a bisphosphonate) and atorvastatin (a cholesterol-lowering drug) decreased MPO activity in the gingiva of rats following ligature-induced periodontitis. 248 Also simvastatin (another inhibitor of 3-hydroxy-3-methylglutaryl-CoA reductase, the key enzyme in endogenous cholesterol biosynthesis pathway), which was previously reported to downregulate MPO gene expression in human and murine monocyte macrophages, 249 was found to decrease MPO activity dose dependently in experimental periodontitis. 250 Additionally, a recent study in patients with type-2 diabetes with hypercholesterolemia demonstrated the mechanisms of lipid-lowering drugs to reduce systemic pro-inflammatory factors and MMPs. 251
Inhibition of MPO or scavenging of HOCl might represent an alternative strategy for the treatment of PDs. 252 In particular, taurine chloramine, which is generated from HOCl and its physiological scavenger (the sulfur-containing aa taurine), is only about a third as active as HOCl in activating MMP-8 but completely fails to inhibit TIMP-1 at concentrations achieved at sites of inflammation. 253
Conclusion
A recent study correlating salivary biomarkers with MI and periodontal status revealed that MMP-9 correlated positively with MMP-8 and MPO but negatively with TIMP levels. 254 Indeed, MMP-8 and MMP-9 primarily secreted from neutrophils in a latent form and during inflammation can be activated by several pro-inflammatory mediators, such as cytokines, MMP-7, NE and MPO-derived/generated reactive intermediates (including HOCl), as well as microbial proteases. 99 MMP-8 is able to degrade collagen type I, the major contributor to the tensile strength of the fibrous cap, threefold more potently than other interstitial collagenases.78,81 In addition to BM proteolysis, the proteolytic products of collagens can further be degraded by MMP-9. 71 Through its ability to release chemokines, epithelial MMP-7 is important for neutrophil influx to the site of inflammation.124,255 Other enzymes of the neutrophil granules, for example NE and MPO, and MPO-generated reactants, secreted into the extracellular space can cleave the ECM. MMPs and their regulators can promote auto-activation and form proteolytic activation cascades. 108 Alternatively, MPO potentiates MMP proteolytic cascades by impairing the crucial balance between proteases and anti-proteases that may lead to potentially deleterious situations. 256 MPO can also oxidatively convert latent MMP-8 and MMP-9 into proteolytically active forms, inactivate TIMPs and regulate neutrophil recruitment (Figures 1 and 2).69,76,101,190,257 All of these neutrophil-derived markers, reported to be upregulated in atherosclerotic lesions, are thought to play a fundamental role in plaque rupture and are associated with subsequent pathological CVD events.93,94,148,168,258,259 Thus, neutrophil-derived proteases implicated in the atherosclerotic plaque rupture may lead to acute manifestations of the disease such as unstable angina pectoris or acute MI. On the one hand, future CVD events could be predicted by determining serum MMP-7 and MMP-8, TIMP-1 and the MMP-8/TIMP-1 ratio, as high MMP-7 and MMP-8 levels have been associated with several forms of CVD and increased incidence of fatal heart attacks.92–96 Serum levels of these molecules might reflect the progression and severity of CVD and may thus be considered as candidate markers in predicting future CVD events. On the other hand, the usefulness of these molecules in oral samples, saliva, mouth rinse or GCF as biomarkers of CVD requires further investigations.
MMP inhibition via doxycycline represents a promising route for periodontitis treatment. Nevertheless, further research is needed that includes large-scale/multinational intervention trials with a sufficiently long follow-up period, standardized periodontal measurements and proper adjustments for known confounders. It is important to elaborate the understanding of molecular mechanisms related to neutrophil proteolytic pathways by investigating the impact of periodontal therapy on traditional CVD risk factors. Furthermore, it is necessary to provide therapeutic strategies preventing and treating severe clinical outcomes, such as secondary CVD events or even mortality. The strategy to balance MMPs and their regulators by doxycycline treatment might offer a suitable approach for CHD treatment by providing an anti-proteolytic and anti-inflammatory barrier against systemic inflammation and recurrent CVD events.
Footnotes
Funding
Research in our laboratories has been funded by the Ella and Georg Ehrnrooth Foundation (to HA), the Sigrid Juselius Foundation (to PJP), the Yrjö Janhsson Foundation (to PJP), the Finnish Dental Society Apollonia (to PJP), the Academy of Finland (#1266053 to PJP, #1130408 to TS) and the Helsinki University Central Hospital Foundation (EVO for TS).
Conflict of interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.