
Abstract
Over the last 40 yr, the majority of research on glucans has focused on β-(1→3)-glucans. Recent studies indicate that β-(1→6)-glucans may be even more potent immune modulators than β-(1→3)-glucans. Mechanisms by which β-(1→6)-glucans are recognized and modulate immunity are unknown. In this study, we examined the interaction of purified water-soluble β-(1→6)-glucans with macrophage cell lines and primary peritoneal macrophages and the cellular and molecular consequences of this interaction. Our results indicate the existence of a specific β-(1→6)-glucan receptor that internalizes the glucan ligand via a clathrin-dependent mechanism. We show that the known β-(1→3)-glucans receptors are not responsible for β-(1→6)-glucan recognition and interaction. The receptor–ligand uptake/interaction has an apparent dissociation constant (KD) of ∼4 µM, and was associated with phosphorylation of ERK and JNK but not IκB-α or p38. Our results indicate that macrophage interaction with β-(1→6)-glucans may lead to modulation of genes associated with anti-fungal immunity and recruitment/activation of neutrophils. In summary, we show that macrophages specifically bind and internalize β-(1→6)-glucans followed by activation of intracellular signaling and modulation of anti-fungal immune response-related gene regulation. Thus, we conclude that the interaction between innate immunity and β-(1→6)-glucans may play an important role in shaping the anti-fungal immune response.
Introduction
Glucans are structurally diverse fungal biopolymers that stimulate innate immunity.1–3 Fungal β-(1→3)-glucans are generally recognized as important PAMPs in the anti-fungal immune response.4–6 The binding of β-(1→3)-glucans by specific receptors results in activation of anti-fungal innate and adaptive immune responses,3,7–13 leading to effects of β-(1→3)-glucans as immune modulators.14–17 Over the last 40 yr, the vast majority of research on glucans has focused on β-(1→3)-glucans.1–16 However, there are glucans other than β-(1→3)-linked glucans present in the fungal cell wall, although they are generally present at lower levels.18,19 Recent studies suggest that β-(1→6)-glucans may be even more potent immune modulators than β-(1→3)-glucans.20–23 Compared with β-(1→3)-glucans, the β-(1→6)-glucans elicit a more potent induction of IL-1β, IL-6, IL-8 and TNF-α in human whole blood in vitro. 23 Abs directed against β-(1→6)-glucan have been detected in human sera, and the human Ab response to β-(1→6)-glucan was greater than that observed for β-(1→3)-glucan.21,22 In addition, when compared with beads coated with β-(1→3)-glucans, a more efficient internalization of beads coated with β-(1→6)-glucans by neutrophils has been reported. 20
Activation of transcription factors induced by the interaction of β-(1→3)-glucans with immune cells,3,24 as well as up- and/or down-regulation of specific mRNAs,25,26 has been described previously. It is reasonable to speculate that the reported cytokine induction and Ab responses to β-(1→6)-glucans would be preceded by activation of transcription factors and changes in mRNA levels;21–23 however, these changes in cellular and molecular function have not yet been investigated for pure β-(1→6)-glucans.
Dectin-1 has been identified as the principal pattern recognition receptor for β-(1→3)-glucans,8,27 and is expressed on monocytes, macrophages, dendritic cells and neutrophils.27,28 Adams et al. and Palma et al. have reported that Dectin-1 does not recognize β-(1→6)-glucans.7,29 This suggests that there is another receptor that mediates responses to β-(1→6)-glucan. There are three other receptors that have been reported to bind β-(1→3)-glucans: the type 3 complement receptor (CR3),10,30 scavenger receptor A (SRA) and a lactosylceramide,9,11 although these receptors bind β-(1→3)-glucans less selectively than Dectin-1. Zimmermann et al. have reported that lactosylceramide does not bind β-(1→6)-glucans. 11 However, CR3 and SRA have not been evaluated for their ability to interact with β-(1→6)-glucans. Thus, the mechanisms by which β-(1→6)-glucans are recognized and modulate immunity in humans and animals are unknown.
In this study, we investigated the mechanisms associated with the induction of biological activities in macrophages by β-(1→6)-glucans. By employing macrophage cell lines and peritoneal macrophages derived from wild type (WT), Dectin-1 (Dectin-1−/−), SRA−/− or CR3−/−) knockout mice, we explored the recognition and binding specificity of β-(1→6)-glucans by macrophages and what, if any, role known β-(1→3)-glucan receptors may play in this interaction. Additionally, we examined the binding characteristics and mechanisms of internalization of β-(1→6)-glucans by macrophages, as well as the activation of transcription factors and modulation of gene transcription following the interaction between β-(1→6)-glucans and macrophages.
Materials and methods
Ethics statement
All animal procedures were conducted in strict compliance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. The animal protocol was reviewed and approved (protocol number 101201) by the University Committee on Animal Care at the James H. Quillen College of Medicine, East Tennessee State University (ETSU), under the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care, US Department of Agriculture, and the Public Health Service guidelines for the care and use of animals as attested by the National Institutes of Health. All efforts were made to minimize suffering.
Reagents
Alexa Fluor 647 (AF647), propidium iodide and a Vybrant Lipid Raft Labeling Kit were purchased from Molecular Probes (Eugene, OR, USA). DMSO, methyl-β-cyclodextrin, chlorpromazine, nystatin, cytochalasin D and hypotonic sucrose were purchased from Sigma Chemical (St. Louis, MO, USA). Wortmannin was purchased from Alomone laboratories (Jerusalem, Israel). Cytochalasin D and wortmannin were pre-dissolved in DMSO and diluted to the stock concentrations in RPMI, while all other inhibitors were dissolved in serum-free RPMI medium. All stock solutions were incubated overnight in pre-washed polymyxin-coated agarose beads (BioRad, Hercules, CA, USA) to remove endotoxin, and filter sterilized prior to use.
Polysaccharides
Glucan phosphate, a water-soluble β-(1→3)-glucan, was extracted from Saccharomyces cerevisiae, 31 and had a molecular mass of 1.5 × 105 g/mol. Water-soluble β-(1→6)-glucan was isolated from Malassezia sympodialis as previously described, and had a molecular mass of 6 × 104 g/mol. 32 The chemical structures of the glucans used in this study were confirmed by 1H-NMR 33 and the physicochemical characteristics were confirmed as described earlier.3,34 The β-(1→6)-glucan was derivatized with diaminopropane at the reducing terminus and labeled with AF647 as previously described.35,36
Macrophage cell lines and isolation of murine primary macrophages
Human macrophage cell line U-937 (CRL-1593.2), murine macrophage cell lines J774A.1 (TIB-67) and RAW 264.7 (TIB-71) were obtained from American Type Culture Collection (Rockville, MD, USA). Cell lines were grown either as suspension cultures (U937) or adherent monolayers (J774A.I and RAW 264.7) at 37℃ and 5% CO2 in RPMI 1640 (Cellgro, Manassas, VA, USA) containing 9% calf serum, 1% FBS and 1% (v/v) penicillin–streptomycin. Peritoneal macrophages were elicited using thioglycollate and isolated from WT, Dectin-1−/−, SRA−/− or CR3−/− mice on the C57BL/6 J background as previously described. 36 Dectin-1−/− and SRA−/− mice were obtained from breeding colonies in the Department of Laboratory Animal Resources at ETSU. WT and CR3−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The animals were maintained on standard laboratory chow and water was available ad libitum with a 12-h light/dark cycle. Serologic testing confirmed that the mice were virus free. All animal procedures were reviewed and approved by the institutional review board at the James H Quillen College of Medicine, ETSU.
Flow cytometry
Optimal concentrations for measurements of the fluorescent labeled β-(1→6)-glucan in flow cytometry and confocal microscopy were identified in pilot experiments (not shown). Different inhibitor concentrations were tested for their effect on cell viability using the trypan blue exclusion test (not shown). Concentrations were selected to maintain cell viability during treatment. To perform the competition experiments, cells were plated in six-well plates at 3 × 105 cells per well and grown for 48 h at 37℃ in 5% CO2. Peritoneal macrophages were counted and seeded at 6 × 106 cells per well. They were allowed to adhere overnight (12 h). Before treatment, cells were washed and fresh medium was added. Cells were pre-incubated for 30 min with 125 or 500 µg/ml of β-(1→6)-glucan or β-(1→3)-glucan prior to addition of 10 µg/ml fluorescently labeled β-(1→6)-glucan and incubated for another 1.5 h. Untreated cells served as negative control and the positive control only received the 10 µg/ml fluorescently labeled β-(1→6)-glucan. To investigate the internalization pathway, cells were pre-treated with serum-free medium alone (negative and positive control), serum-free medium containing 12.5–50.0 μM nystatin, 2.5–10.0 mM methyl-β-cyclodextrin, 100–400 nM wortmannin, 5–20 μM cytochalasin D, 10–40 μM chlorpromazine or 100–400 µM sucrose for 30 min at 37℃. After pre-treatment, 10 µg/ml of fluorescently labeled β-(1→6)-glucan was added to each well, except to the negative control, and the cells were incubated for an additional 1.5 h at 37℃. For the saturation experiments, WT peritoneal macrophages were counted and seeded at 6 × 106 cells per well. They were allowed to adhere overnight. Before treatment, the cells were washed and fresh medium was added. Cells were incubated with a dilution series of non-labeled β-(1→6)-glucan (0.065–8.300 μM) for 30 min before the addition of 10 µg/ml fluorescently labeled β-(1→6)-glucan. The negative control was left untreated and a positive control was pre-incubated in the absence of competitors but 10 µg/ml fluorescently labeled β-(1→6)-glucan was added. After each experiment, cells were harvested with lidocaine, washed with PBS and re-suspended in Pharmingen Stain Buffer (BD Biosciences, San Jose, CA, USA). Mean fluorescence was measured and analyzed using a FACScalibur flow cytometer with CellQuest software (BD Biosciences).
Confocal microscopy
For confocal microscopy, 2.5 × 105 cells were seeded on sterile cover slips in a 12-well plate and allowed to grow for 48 h. If applicable, cells were pre-treated for 30 min in serum-free medium containing the specified endocytosis inhibitors before addition of 100 µg/ml fluorescently labeled β-(1→6)-glucan. The negative control was left untreated and the positive control was pre-incubated in the absence of inhibitors but did receive 100 µg/ml fluorescently labeled β-(1→6)-glucan. After a 3-h incubation, cover slips were carefully washed with PBS. To visualize the cell membrane, cells were stained with the Vybrant Lipid Raft Labeling Kit. After another washing cycle, cells were fixed with 4% paraformaldehyde. Cover slips were mounted with Prolong Antifade (Molecular Probes). The slides were imaged on a Leica DM IRBE inverted microscope with the Leica TC2 SP2 confocal microscopy system (Leica, Wetzlar, Germany). The images were analyzed using Leica confocal software.
Western blots
For protein measurements, J774a.1 cells were cultured to confluency in T75 flasks. Cells were treated for 15, 30, 60 and 120 min with 100 µg/ml β-(1→6)-glucan. Untreated cells served as control. After treatment, cells were incubated for 1 h in lysis buffer containing 10 mM Hepes (pH 7.9), 10 mM KCL, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 1 μM aprotinin, 14 μM leupeptin, 1 μM pepstatin and 80 µg/ml benzamidine, and then lysed by adding 10% Nonidet P40. Protein concentrations in the cell extracts were quantified with the BCA protein assay (Bio-Rad, Hercules, CA, USA). Fixed amounts of total protein (100 µg for JNK and 50 µg for ERK, IkB-α and p38) were resolved by SDS-PAGE and transferred to PVDA membranes. The membranes were blocked with Tris-buffered saline (TBS) + 0.05% Tween20 + 5% Milk (TBTM) and incubated with rabbit anti-phospho-ERK, rabbit anti-phospho-JNK, rabbit-anti-phospho-p38 or mouse-anti-phospho-IκB-α (Cell Signaling Technology. Beverly, MA, USA) overnight. After washing three times for 10 min in TBTM, membranes were incubated with the corresponding peroxidase-conjugated anti-mouse or peroxidase-conjugated anti-rabbit secondary Abs (Cell Signaling Technology). After washing three times for 10 min in TBS + 0.05% Tween20, membranes were developed by chemiluminescence (Immun-Star WesternC Chemiluminescent Kit; Bio-Rad) and then visualized using a GBox (Syngene). The membranes were washed, re-blocked and re-probed overnight with the appropriate Ab: rabbit anti-ERK, rabbit anti-JNK, rabbit anti-p38 and rabbit anti-IκB-α (Santa Cruz Biotechnology, Inc., CA) followed by incubation with peroxidase-conjugated secondary anti-rabbit Abs (Sigma Chemical, MO). Membranes were developed and imaged as before. Next, membranes were stripped and re-probed with anti-GAPDH (Biodesign, ME) as loading control, followed by incubation with peroxidase-conjugated secondary anti-mouse Abs (Sigma Chemical, MO). Membranes were developed by chemiluminescence and visualized using a GBox.
PCR Array
The PCR array procedure was performed following the protocol supplied with the RT 2 Profiler PCR Array (Qiagen). Briefly, J774a.1 cells were cultured to 80% confluency in T75 flasks. Cells were treated for 1, 3, 6, 12 and 24 h with 100 µg/ml β-(1→6)-glucan. Controls were left untreated. Total RNA was extracted using the RNeasy mini kit (Qiagen) and purity was controlled using an Agilent Bioanalyzer. The cDNA was prepared using the RT 2 First Strand Kit (Qiagen), the sample was then mixed with RT 2 SYBR Green Master mix and the real time PCR program was run according to the manufacturer’s specifications.
Data analysis
All data was corrected for the appropriate negative controls.
Competition data is presented as the percent fluorescence relative to the fluorescence of the corresponding positive controls. Receptor binding/uptake versus concentration data were normalized for fluorescence based on the proportion of total to labeled glucan. Corrected fluorescence values were fit to a single-site hyperbolic binding model using nonlinear least squares regression with GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, CA) to calculate an apparent KD and Bmax for the receptor binding/uptake process. Western blot results were analyzed by measuring the fluorescent intensity of the negatives of the imaged membranes using GeneTools (Syngene, Frederick, MA, USA). Values were normalized to a GAPDH loading control before calculating up-regulation of phosphorylation. Up- or down-regulation was calculated comparing the phospho-protein to the total protein (fold regulation). All experiments were repeated at least three times. Groups were compared by ANOVA applying post-hoc testing using Tukey’s procedure; probability levels of P ≤ 0.05 were considered significant.
The PCR array experiment was repeated twice and analyzed using the Qiagen PCR Array Data Analysis Web portal. Up- or down-regulation of mRNAs was calculated by comparing treated samples with non-treated controls (fold regulation). Significance for up- or down-regulation was set to a difference in fold regulation of at least two. Figures and statistic calculations for flow cytometry experiments and Western blot analysis were produced in Sigmaplot 10 (Systat Software, San Jose, CA, USA), if not otherwise specified. Figures for the PCR array data were produced in with GraphPad Prism (GraphPad, La Jolla, CA, USA).
Results
Specific binding and internalization of β-(1→6)-glucan by murine macrophages
Internalization of fluorescently labeled β-(1→6)-glucan was explored using three different macrophage cell lines to exclude possible artifacts due to one specific cell line (Figure 1A). Initially, we employed flow cytometry and confocal microscopy to confirm that all three cell lines bound and internalized β-(1→6)-glucan (data not shown). Once we confirmed that all three cell lines interacted with the glucan, we examined the specificity of this interaction with competition experiments using a 50-fold excess of non-labeled β-(1→6)-glucan or non-labeled β-(1→3)-glucan (Figure 1A). To demonstrate concentration dependency we employed a 12.5- and 50-fold excess of non-labeled glucan in J774a.1 cells (Figure 1B). Specificity of the interaction between the macrophage cell lines with β-(1→6)-glucan was clearly demonstrated. A significant reduction in binding/internalization of 20–30% was observed for the higher concentration (500 µg) of non-labeled β-(1→6)-glucan (Figure 1A). The 125 µg competitor concentration of β-(1→6)-glucan inhibited the interaction with macrophages by 10–20% (Figure 1B). In contrast, non-labeled β-(1→3)-glucan did not inhibit the interaction of labeled β-(1→6)-glucan with the macrophages. Interestingly, the lower concentration (125 µg/ml) of β-(1→3)-glucan, when used as a competitor, seems to augment the binding/internalization of labeled β-(1→6)-glucan (Figure 1B).
Specific binding and internalization of β-(1→6)-glucan by macrophage cell lines. (A) Human U937 and murine RAW and J774a.1 cell lines were treated with 50 × higher concentration of non-labeled competitors—β-(1→6)-glucan or β-(1→3)-glucan—prior to the treatment with 10 µg/ml (167 nM) labeled β-(1→6)-glucan. (B) J774a.1 cells treated with 12.5×or 50 × higher concentrations of non-labeled competitors—β-(1→6)-glucan or β-(1→3)-glucan—prior to the treatment with 10 µg/ml (167 nM) labeled β-(1→6)-glucan. The data are presented as the percent fluorescence relative to the fluorescence of the corresponding positive controls ± SD of four repeated measurements per group. *Significant difference compared with control (P ≤ 0.05).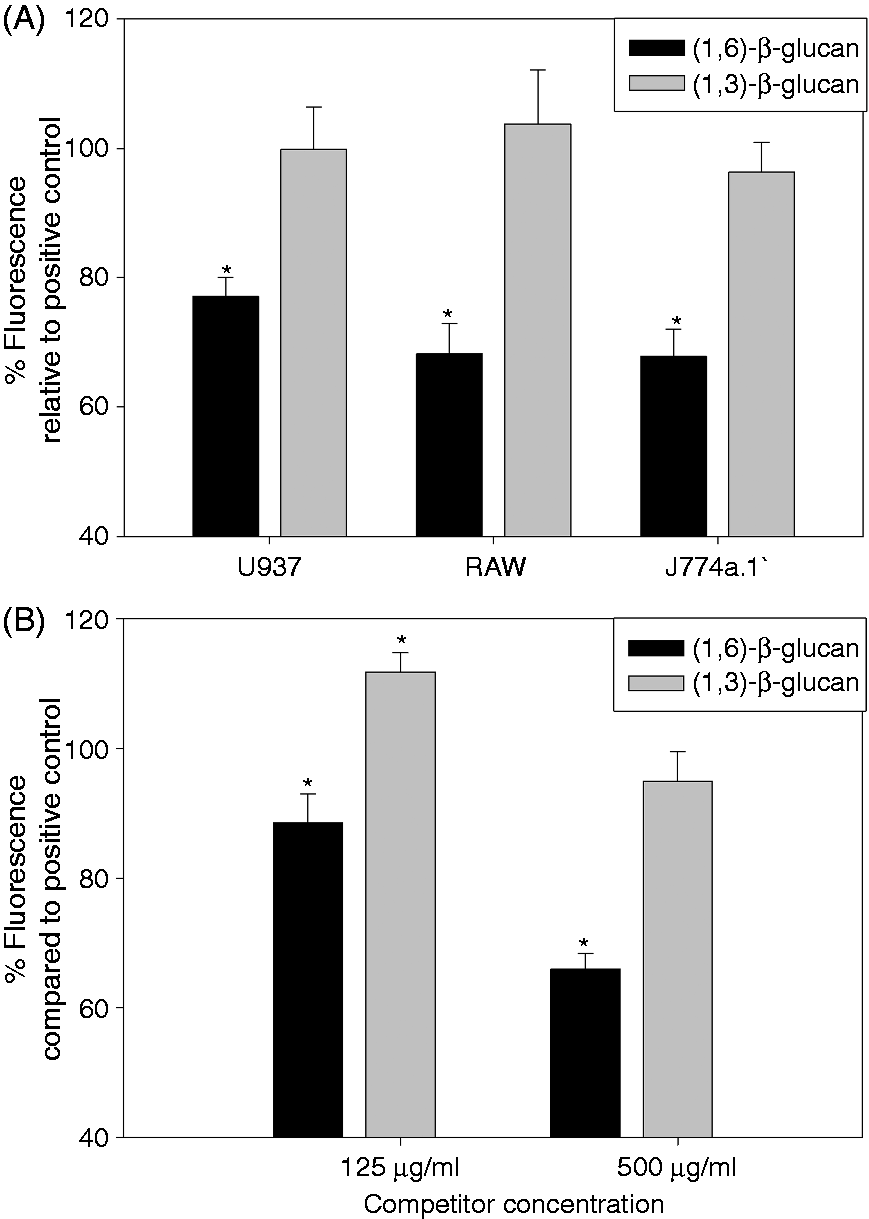
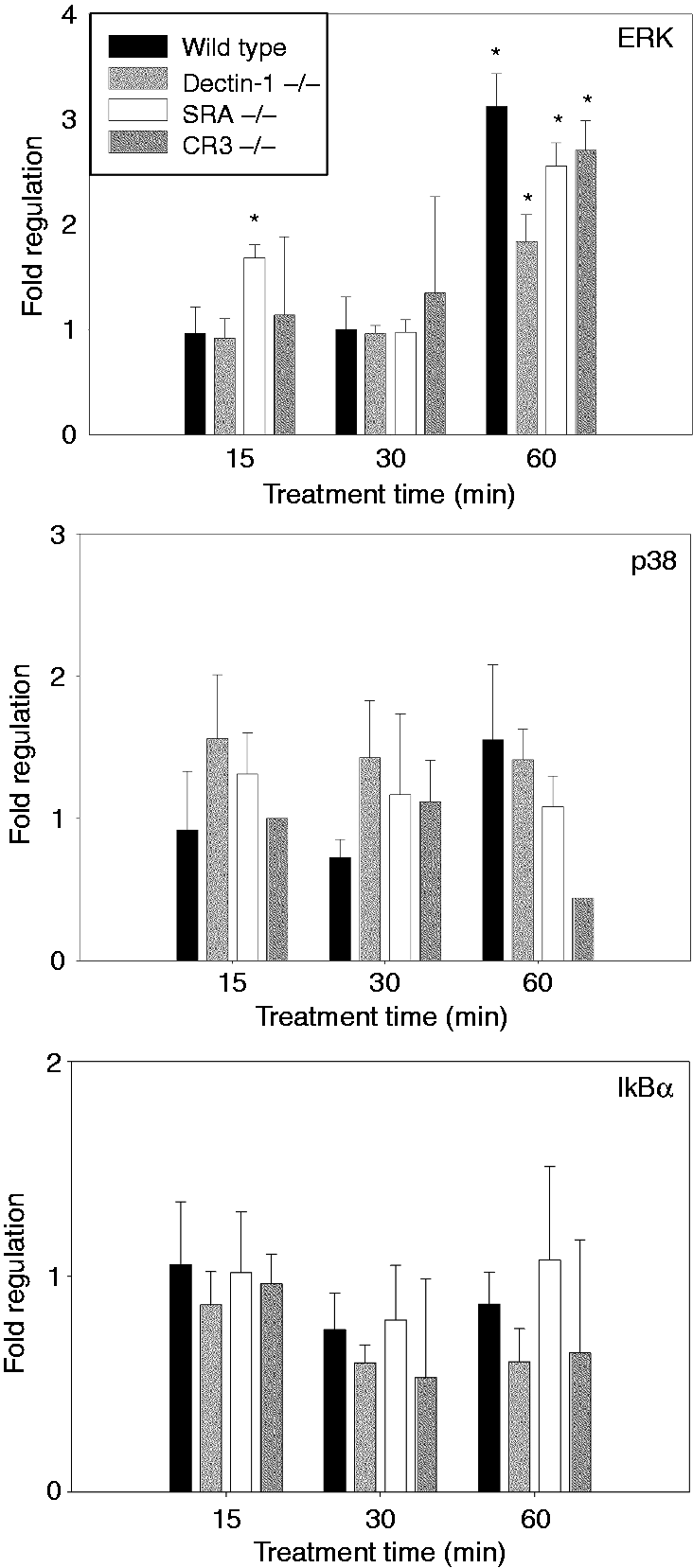
Uptake of β-(1→6)-glucan macrophages is not mediated by Dectin-1, SRA or CR3
In order to investigate whether the binding and internalization of β-(1→6)-glucan is dependent on one of the previously reported β-(1→3)-glucan receptors,9–11,30 we harvested peritoneal macrophages from mice that are genetically deficient for the respective receptors (Figure 2). The results of the competition experiments using primary macrophages were more dramatic than those observed in cell lines, but similar trends were observed. A 50-fold excess of non-labeled β-(1→6)-glucan reduced the binding/internalization of labeled β-(1→6)-glucan by 50–60% (P < 0.05) in thioglycollate-induced macrophages derived from all the mice strains employed (Figure 2). Thus, primary macrophages derived from WT and receptor knockout mice produced similar results, indicating that the receptors investigated, that is, Dectin-1, SRA or CR3, do not participate in the binding or internalization of β-(1→6)-glucan.
Specific binding and internalization of β-(1→6)-glucan by murine primary macrophages. Macrophages were treated with unlabeled competitors—β-(1→6)-glucan or (β-(1→3)-glucan—prior to the treatment with labeled β-(1→6)-glucan. The data are presented as the percent fluorescence relative to the fluorescence of the corresponding positive controls ± SD of four repeated measurements per group. *Significant difference compared with controls without competitor (P ≤ 0.05).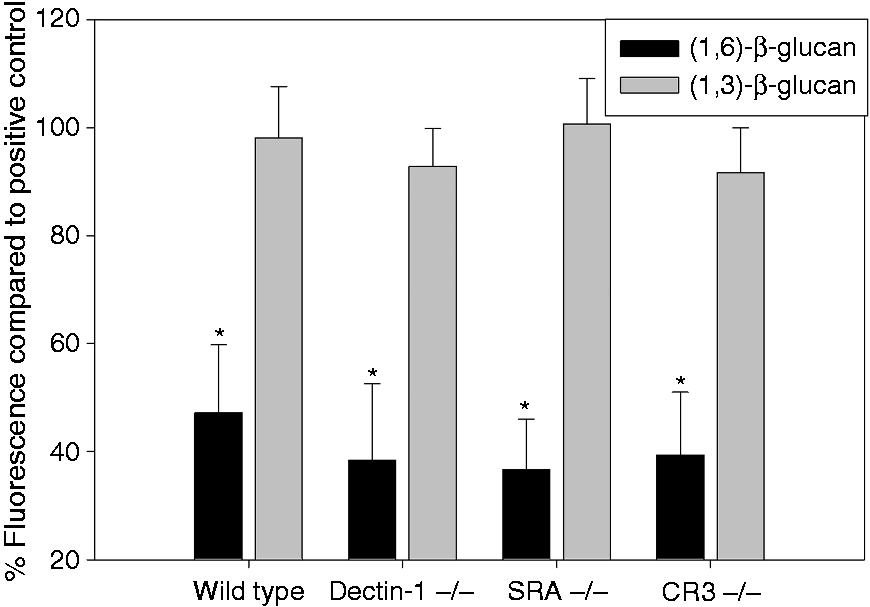
β-(1→6)-Glucan is internalized by macrophages via a clathrin-dependent endocytosis mechanism
As we could demonstrate specific binding and internalization of β-(1→6)-glucan by monocytes/macrophages, we investigated the mechanisms of β-(1→6)-glucan internalization. We treated cells with two known inhibitors of each of three distinct internalization pathways: the lipid raft-mediated endocytosis pathway, phagocytosis and clathrin-mediated endocytosis. Quantitative changes in binding and internalization of fluorescently labeled β-(1→6)-glucan were analyzed using flow cytometry (Figure 3A). Inhibitors of lipid raft-mediated endocytosis, nystatin and methyl-β-cyclodextrin did not inhibit binding and/or internalization of the β-(1→6)-glucan. Wortmannin, a phagocytosis inhibitor, did not show significant inhibition; however, cytochalasin D at 20 µM significantly inhibited binding and internalization of β-(1→6)-glucan by 20–40%. Inhibitors of clathrin-mediated endocytosis showed the most dramatic effect by significantly inhibiting binding and internalization of β-(1→6)-glucan. Chlorpromazine (40 µM) inhibited glucan uptake by 20–40%, while hypotonic sucrose (100 mM) inhibited internalization of β-(1→6)-glucan by 25–45% and by 80% at 400 mM. We confirmed these observations qualitatively using suitable inhibitors and confocal microscopy (Figure 3B). In agreement with the flow cytometry data, methyl-β-cyclodextrin and wortmannin did not inhibit the binding/internalization of the fluorescently labeled β-(1→6)-glucan, while chlorpromazine treatment clearly reduced bound/internalized fluorescently labeled β-(1→6)-glucan.
Internalization of β-(1→6)-glucan by macrophages is mediated via a clathrin-dependent mechanism. Various inhibitors specific for the different pathways were used. (A) Data as measured by flow cytometry. The data are presented as the percent fluorescence relative to the fluorescence of the corresponding positive controls ± SD of four repeated measurements per group. (B) Data as measured by confocal microscopy. *Significant difference compared with controls without competitor (P ≤ 0.05).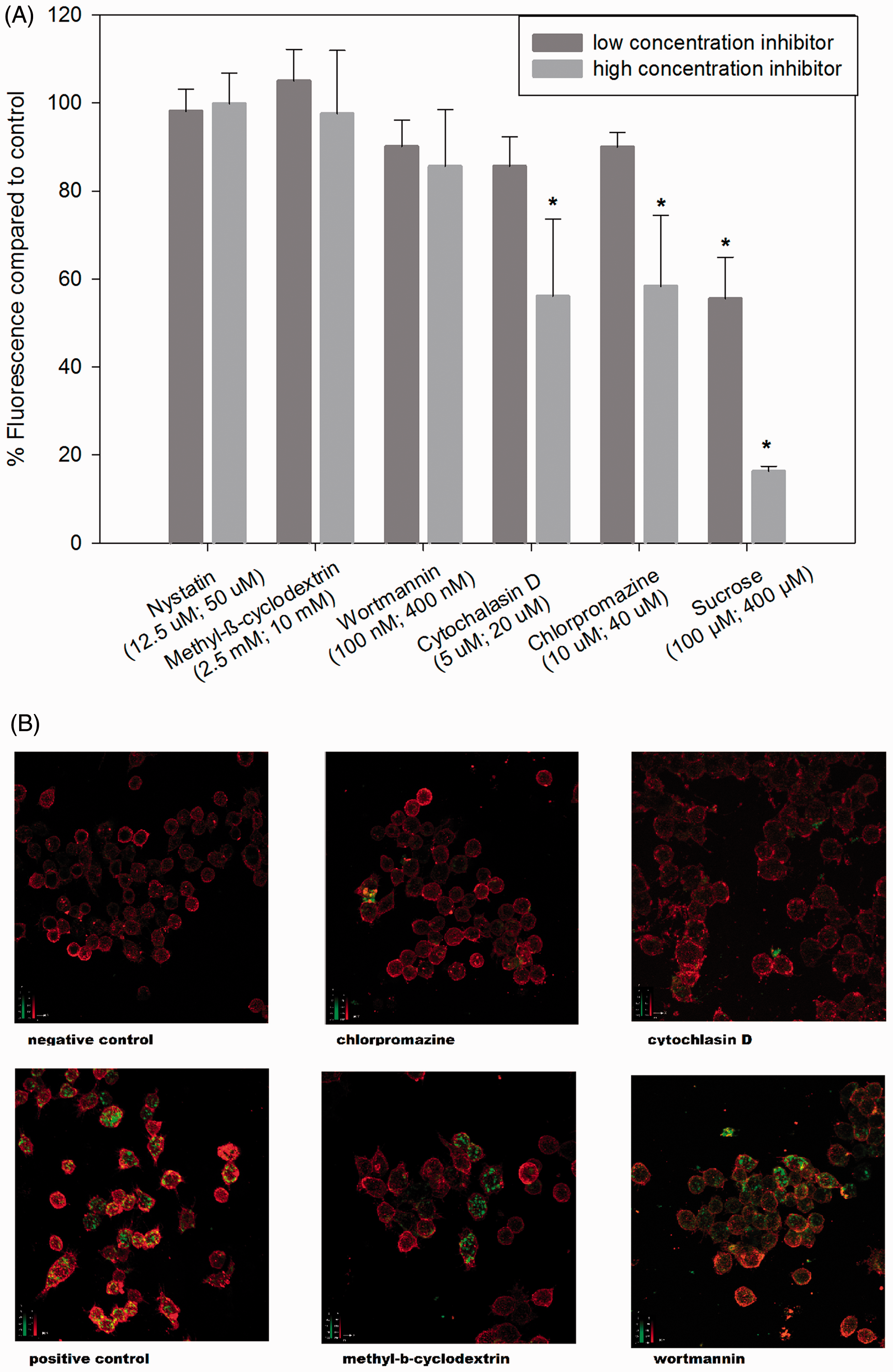
β-(1→6)-Glucan binds to murine peritoneal macrophages in a saturable, dose-dependent manner
Our results strongly indicate a receptor-mediated binding and internalization of β-(1→6)-glucan, which occurs in a specific manner. While we have not yet identified the receptor, we investigated the binding kinetics of the interaction between β-(1→6)-glucan and its putative receptor in murine primary peritoneal macrophages. Using a single site-specific binding model, fitting combined saturation and competitive displacement data yielded an apparent dissociation constant (KD) of 4 µM [95% confidence interval (CI) 2.5–5.0 µM) and a maximum fluorescence (Bmax) of ∼61 fluorescence units (FU)/cell (95% CI 50.0–71.0 FU/cell) (Figure 4).
Saturability and dose dependence of β-(1→6)-glucan in primary macrophages. Data from three replicate experiments were fitted to a single-site hyperbolic binding model using nonlinear least squares regression. Combined saturation and competitive displacement data yielded a KD of 4 µM and a Bmax of 61.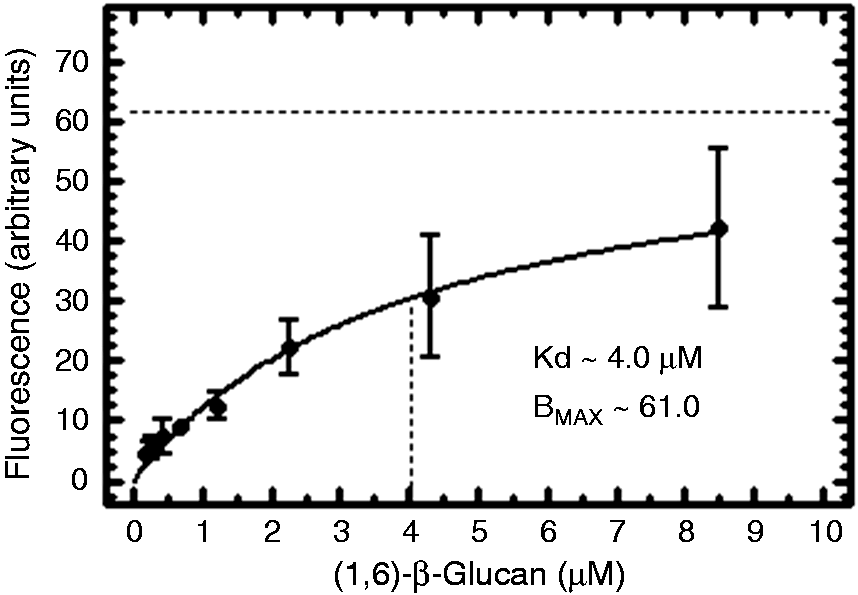
β-(1→6)-Glucan activates the MAPKs, ERK and JNK but not p38 or NF-κB
To investigate possible activation of signal transduction, we measured IκB-α phosphorylation as an index of NF-κB activation and the phosphorylation of the MAPKs ERK, JNK or p38 in β-(1→6)-glucan-treated J774a.1 cells (Figure 5A, B). Phosphorylation was significantly up-regulated at 15 min (∼15 × higher than control) and 30 min (∼3 × higher than control) for ERK and at 15 min (∼2 × higher than control) for JNK (Figure 5B). There was no significant difference in phosphorylation status of p38 or IκB-α in the J774a.1-treated cells when compared with the non-treated control at any time point. In peritoneal macrophages (Figure 6), we observed a similar up-regulation of ERK, although the kinetics of the response was different in that peak activity was observed at 60 min, as opposed to 15 min (Figure 5). As seen in the J774a.1 cells, p38 and IkB-α showed no change. We were not able to detect total or phosphorylated JNK in primary peritoneal macrophages at any time point in the primary cells.
Effect of β-(1→6)-glucan on phosphorylation of IκB-α, ERK, JNK and p38 in murine macrophage cell line J774a.1. (A) Representative results of membranes being probed for phosphorylated protein, re-probed for total protein, stripped and re-probed for loading control. (B) Fold regulation (ratio phosphorylated to total protein) of phosphorylation status of the measured proteins at different time points. Data are expressed as the ratio of phospho-protein to total protein (fold regulation) ± SD of three repeated measurements. *Significant difference when compared with non-treated control (P ≤ 0.05). Phosphorylation status of IκB-α, ERK and p38 in primary peritoneal macrophages treated with β-(1→6)-glucan. The data are presented as fold regulation (ratio of phosphorylated to total protein) of phosphorylation status of the measured proteins at different time points. Data are expressed as the ratio of phospho-protein to total protein (fold regulation) ± SD of three repeated measurements. *Significant difference when compared with non-treated control (P ≤ 0.05).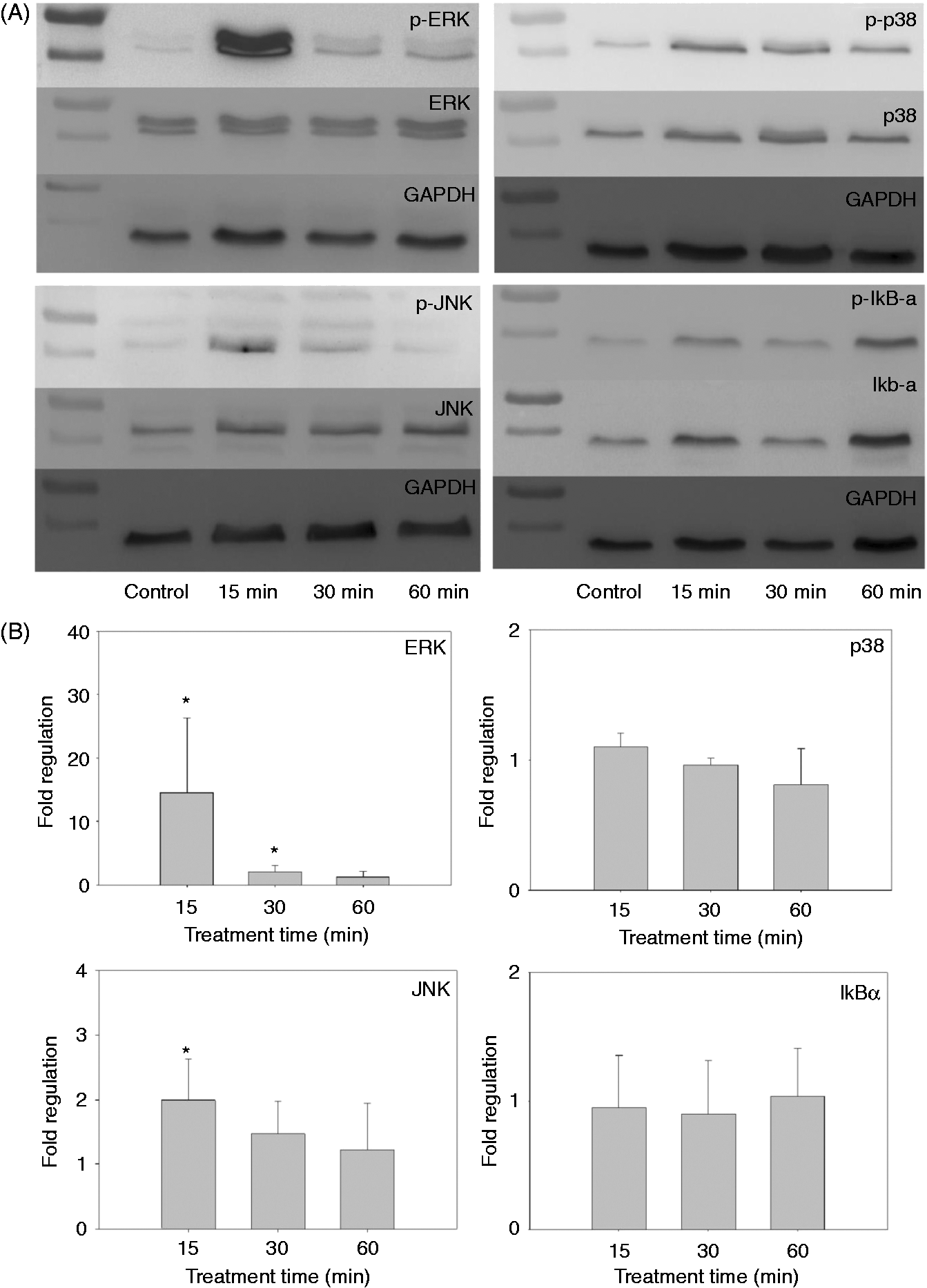
Macrophage activation by β-(1→6)-glucan results in modulation of genes specific for anti-fungal immune responses
The activation of a signal transduction cascade leads to changes in transcription factor activity and induces changes in gene transcription, that is, mRNA levels. We used PCR array technology to measure changes in mRNA levels of genes important for anti-fungal immune responses in J774a.1 cells after treatment with β-(1→6)-glucan at different time points (Figure 7A, B). J774a.1 cells were treated with β-(1→6)-glucan for 1, 3, 6, 12, and 24 h. Three mRNAs were significantly up-regulated (≥ 4 ×) at various time points: C-X-C motif chemokine 10 (Cxcl10), complement component 5a receptor 1 (C5ar1) and coagulation factor III (F3). Ten mRNAs were significantly down-regulated [≥ 4 × s: prostaglandin-endoperoxide synthase 2 (Ptgs2), IL-1β (Il1β), Il23α, Il6, IL-1 receptor type 1 (Il1r1), CD83, transcription factor Fos, C-C motif chemokine receptors type 1 and 5 (Ccr1, Ccr5) and chemokine C-X-C motif ligand 3 (Cxcl3)]. Fcgr1 and CD40 were down-regulated (2–3-fold), while Tnf, Pycard, Socs3, Ptpn6, Scarf1, Csf2 and Csf3 were up-regulated 2–3-fold.
Treatment of the macrophage cell line J774a.1 with β-(1→6)-glucan results in (A) up- or (B) down-regulation of mRNAs associated with anti-fungal immune responses. Data are expressed as the ratio of treated samples to non-treated controls (fold regulation) of two repeated measurements. *Significance based on ≥ two-fold changes; **significance based on ≥ four-fold changes as suggested by the manufacturer.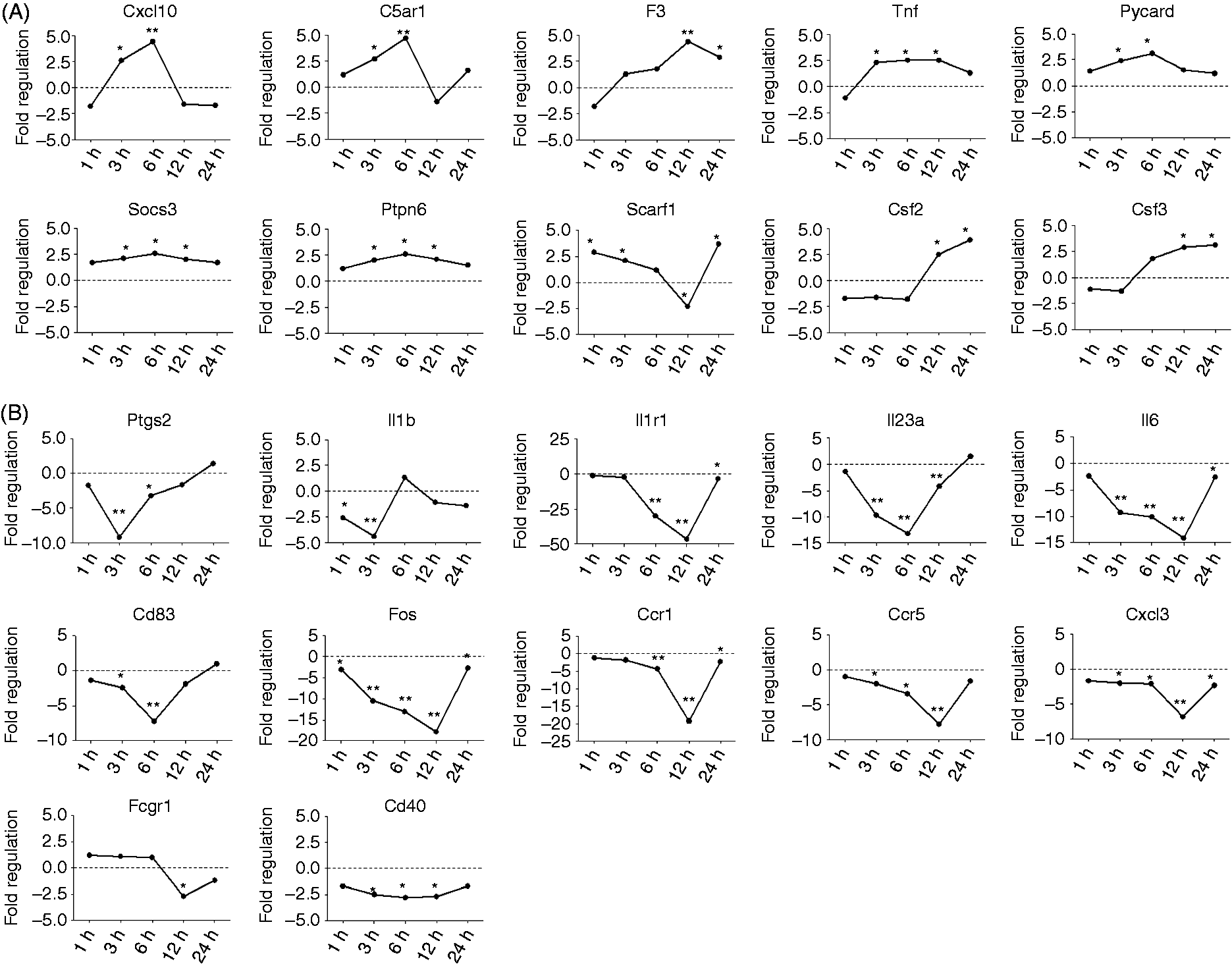
Discussion
We hypothesized that β-(1→6)-glucans are recognized by (an) as yet unidentified receptor(s) in the mammalian innate immune system and that this interaction will lead to modulation of innate immunity. Using human and mouse macrophage cell lines we observed that unlabeled β-(1→6)-glucan, but not β-(1→3)-glucan, competed for binding/internalization with labeled β-(1→6)-glucan. This indicates a specific interaction between the macrophages and β-(1→6)-glucan.
The apparent augmentation of the binding/internalization of labeled β-(1→6)-glucan by 125 µg/ml of β-(1→3)-glucan, when used as a competitor, may point to a dynamic interplay between the different glucan types at certain concentrations and needs further investigation.
Employing peritoneal macrophages derived from WT, Dectin-1−/−, SRA−/− or CR3−/− mice, we found that non-labeled β-(1→6)-glucan inhibited the binding/internalization of labeled β-(1→6)-glucan in macrophages from all of the mouse strains. The results were comparable between WT, Dectin-1, SRA and CR3 knockout mice, demonstrating that the binding/internalization of β-(1→6)-glucan is not influenced by any of the known β-(1→3)-glucan receptors. These data are in line with the previous reports that Dectin-1 does not bind β-(1→6)-glucan.7,29 In addition, our data extend these observations to demonstrate that β-(1→6)-glucan is not bound by SRA or CR3.
The uptake of glucans may occur via phagocytosis,37,38 caveolae/lipid raft-mediated endocytosis, 39 which utilizes lipid rafts enriched in sphingolipids such as lactosylceramide, 40 or via clathrin-mediated endocytosis, a pathway by which many receptor-bound ligands are internalized. 41 As inhibitors of these pathways have been shown to sometimes induce diverse effects on the cells, leading to faulty conclusions, 42 we used two structurally dissimilar inhibitors for each pathway. Inhibiting lipid raft-mediated endocytosis did not affect β-(1→6)-glucan uptake. This is supported by the results of Zimmermann et al., 11 which show that lactosylceramide, commonly found in lipid rafts, does not interact with β-(1→6)-glucan. Of the phagocytosis inhibitors, wortmannin inhibiting PI3K did not interfere with internalization of β-(1→6)-glucan. 42 However, cytochalasin D, a blocker of actin polymerization, 42 and both inhibitors of clathrin-mediated uptake abrogated β-(1→6)-glucan internalization. As actin polymerization is necessary for clathrin-mediated endocytosis, 43 the inhibition of β-(1→6)-glucan internalization by cytochalasin D is consistent with the clathrin inhibition data. We conclude that the majority of β-(1→6)-glucan is internalized via clathrin-mediated endocytosis.
We explored the binding kinetics of fluorescently labeled β-(1→6)-glucan and primary murine macrophages. The results yielded an apparent KD of ∼4 µM and a Bmax of ∼61 FU/cell. A KD of 4 µM is rather high and indicates a low affinity for the ligand. Compared with the KD of 24 µM reported for β-(1→3)-glucans using a similar experimental design, 44 the measured KD for β-(1→6)-glucan is lower. Also, Kougias et al., 35 who used fluorescently labeled glucans, reported a KD of 5.6 µM for β-(1→3)-glucan binding to macrophage membranes. Thus, the rather high KD value measured in the present study for β-(1→6)-glucan binding is comparable with the high KD value for receptor binding of β-(1→3)-glucans, which have been generally accepted as important fungal PAMPs.4–6
We investigated the phosphorylation of IκB-α, as a marker for NF-κB activation, and the activation of MAPKs, including ERK, JNK and p38, as these signaling pathways have been reported to be activated by some β-(1→3)-glucans and fungi.24,45 Using the macrophage cell line J774a.1, we found that ERK and JNK phosphorylation were significantly up-regulated, while IκB-α and p38 did not show a change in phosphorylation status. Primary macrophages showed similar results for phosphorylation status of ERK, although ERK phosphorylation kinetics were delayed when compared with the J774a.1 cells. For β-(1→3)-glucans, activation of NF-κB has generally been reported;24,46 thus, our results suggest that β-(1→6)-glucans activate different signal transduction pathways than those reported for β-(1→3)-glucans.
Analyzing the effect of β-(1→6)-glucans on modulation of gene transcription using a commercial PCR array led to the observation that after β-(1→6)-glucan interaction with macrophages, generally more genes were down-regulated than were up-regulated. The mRNAs for the inflammatory mediators IL-1β, IL-6, PTGS2 and IL-1R1 were down-regulated, while the mRNAs for inflammatory mediators TNF-α, PYCARD, SOCS3 and F3 were up-regulated. These changes in the mRNA profile suggest a specific immune response, as opposed to general inflammation. Rand et al. measured gene regulation in alveolar macrophages, 26 and Hida et al. in splenocytes, 25 after treatment with β-(1→3)-glucans. Both studies found an up-regulation of IL-1β and IL-6 mRNAs, while in our study we found that these mRNAs were down-regulated by β-(1→6)-glucan. We did not examine the protein levels of cytokines in this study; however, future investigations should include cytokine expression, as a strong induction of pro-inflammatory cytokine production by β-(1→6)-glucans has been reported in human whole blood. 23
In this study, Cxcl10, C5ar1, Csf2 and Csf3 mRNAs were up-regulated in macrophages treated with β-(1→6)-glucan. These genes code for chemotactic proteins that interact with granulocytes.47,48 In contrast, Cxcl3, Ccr1 and Ccr5 mRNAs, which code for proteins responsible for chemotaxis of leukocytes and especially monocytes, were down-regulated by β-(1→6)-glucan. These results indicate that β-(1→6)-glucans induce a macrophage response that may result in activation and recruitment of neutrophilic granulocytes. This is in line with the results of Rubin-Bejerano et al., 20 who reported that β-(1→6)-glucans coated onto beads efficiently mediated neutrophil activity.
The identification of receptors responsible for recognition and response to fungal pathogens is essential to understanding the mechanisms by which innate immunity responds to these microbes. Our results strongly indicate the existence of a specific receptor for β-(1→6)-glucans on macrophages. Our data exclude several known β-(1→3)-glucan receptors as putative β-(1→6)-glucan receptors. However, thus far, the putative receptor remains unidentified. Nevertheless, the β-(1→6)-glucan interaction with a specific receptor may play an important role in the anti-fungal immune response. Indeed, many studies have reported the importance of the β-(1→6)-glucan side chains in β-(1→3)-glucan bioactivity.49,50 In the current study, we employed a pure β-(1→6)-glucan. Its bioactivity is clearly different from pure β-(1→3)-glucans in activation of transcription factors and regulation of mRNAs. The results indicate the induction of a specific immune response leading to activation and recruitment of neutrophilic granulocytes by β-(1→6)-glucan. This suggests that β-(1→6)-glucan may be an important fungal PAMP that may play a distinct and novel role in shaping the anti-fungal immune response.
Footnotes
Funding
This work was supported, in part, by the German Research Foundation grant NO 928/1-1 to IN, and by National Institutes of Health grant GM53522 and GM083016 to DLW.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.