The AMP-activated protein kinase (AMPK)-mediated energy-sensing signals play important roles in reprogramming the expression of inflammatory genes. In the present study, the potential effects of the AMPK activator 5-aminoimidazole-4 - carboxamide-1-β-D-ribofuranoside (AICAR) were investigated in a mouse model with LPS/D-Gal-induced acute hepatitis. Our experimental data indicated that treatment with AICAR suppressed the elevation of plasma aminotransferases and alleviated the histopathological abnormalities in mice exposed to LPS/D-Gal. Treatment with AICAR also inhibited the LPS/D-Gal-induced up-regulation of TNF-α, NO and myeloperoxidase. In addition, the LPS/D-Gal-induced expression of pro-apoptotic factor Bax, cleavage of caspase-3, elevation of hepatic caspase-3, caspase-8, caspase-9 activities and induction of terminal deoxynucleotidyl transferase-mediated nucleotide nick-end labeling–positive cells were all suppressed by AICAR. These results suggested that the AMPK activator AICAR could attenuate LPS/D-Gal-induced acute hepatitis, which implies that AMPK might become a novel target for the treatment of inflammation-based liver disorders.
LPS, also known as endotoxin, is a major virulence factor of Gram-negative bacteria.1 LPS is a strong stimulator of inflammatory response and is involved in the progression of various infectious and non-infectious diseases.2 Administration of LPS into D-Gal-sensitized mice selectively induces inflammatory liver injury that closely resembles human hepatitis.3 LPS/D-Gal-induced liver injury in mice is a well-established experimental hepatitis model widely used in the investigation of the pathological mechanisms of hepatitis and the development of novel therapeutic approaches for the treatment of hepatitis.4
The essential energy sensor, AMP-activated protein kinase (AMPK), is emerging as an attractive target for pathogenetic investigation and drug discovery.5,6 AMPK is activated under falling energy status, and restores energy and nutrient homoeostasis.7 Accompanied by its modulatory actions on nutrient metabolism, AMPK also participates in the regulation of energy-intensive pathological responses such as inflammation.8,9 It was reported that overexpression of constitutively active AMPK significantly inhibited LPS-induced pro-inflammatory cytokine TNF-α production, whereas the expression of pro-inflammatory genes was enhanced by dominant-negative AMPK or short hairpin RNA targeting AMPK.10,11 Therefore, the AMPK-mediated energy-sensing signals were suggested to be involved in the reprogramming of inflammation.12,13
To investigate the potential roles of AMPK in LPS/D-Gal-induced hepatitis, the potential pharmacological effects of 5-aminoimidazole-4-carboxamide-1-β-D- ribofuranoside (AICAR), a widely used AMPK activator,14,15 was investigated. The administration of AICAR induces the phosphorylation and activation of AMPK, and the therapeutic benefits of AICAR have been previously confirmed in animal model with pneumonia and colitis.16,17 In the present study, AICAR was administrated to a mouse model with LPS/D-Gal-induced liver injury and the degree of liver damage, hepatic inflammation and hepatocyte apoptosis were determined.
Materials and methods
Materials
AICAR, LPS (from Escherichia coli, 055:B5) and D-Gal were purchased from Sigma (St. Louis, MO, USA). The alanine aminotransferase (ALT) and aspartate aminotransferase (AST) assay kits and myeloperoxidase (MPO) assay kit were produced by Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The ELISA kit for detecting mouse TNF-α was from NeoBioscience Technology Company (Shenzhen, China). The total protein extract kit, NO assay kit and caspase-3, caspase-8 and caspase-9 colorimetric assay kits were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). An In Situ Cell Death Detection Kit was purchased from Roche (Indianapolis, IN, USA). The rabbit anti-mouse Bax, Bcl-2 and β-actin Abs were purchased from Cell Signaling Technology (Danvers, MA, USA). The BCA protein assay kit, HRP-conjugated goat anti-rabbit Ab and enhanced chemiluminescence (ECL) reagents were obtained from Pierce Biotechnology (Rockford, IL, USA).
Animals
Male BALB/c mice weighing 20–25 g were obtained from the Experimental Animal Center of Chongqing Medical University. The animals were housed in a specific pathogen-free room at a temperature of 20–25℃ and 50 ± 5% relative humidity under a 12-h dark/light cycle. All animals were fed with a standard laboratory diet and water ad libitum, and acclimatized for at least l wk before use. All experimental procedures involving animals were approved by the Animal Care and Use Committee of Chongqing Medical University.
LPS/D-Gal-induced liver injury
Hepatitis was induced in BALB/c mice by i.p. injection of LPS (10 µg/kg) combined with D-Gal (700 mg/kg) and vehicle or a serial dose of AICAR (100 mg/kg, 200 mg/kg and 300 mg/kg, dissolved in normal saline solution, i.p.) was administrated 0.5 h prior to LPS/D-Gal challenge. Then, the animals were returned to their cages and allowed food and water ad libitum. To evaluate the degree of hepatic lesions, mice were sacrificed 6 h after LPS/D-Gal challenge. The plasma samples were harvested for determining AST and ALT activities. The right lobe of the liver was fixed in formalin for morphological examination and the remaining liver tissues were stored at –80℃ until required. To determine the degree of inflammation, another set of animals was sacrificed at 1.5 h after LPS/D-Gal challenge. The liver and plasma samples were harvested for measuring the mRNA and protein level of pro-inflammatory cytokine TNF-α.
Determination of liver enzymes
The plasma enzyme activities of ALT and AST were assessed according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute).
Histological analysis
Formalin-fixed specimens were embedded in paraffin and stained with hematoxylin and eosin for histopathological evaluation under a light microscope (Olympus, Tokyo, Japan).
TNF-α determination by ELISA
The protein levels of TNF-α in plasma and liver tissue were determined using ELISA kits according to the manufacturer’s instructions (NeoBioscience Technology Company). The levels of hepatic TNF-α were normalized by the total protein concentration of each sample.
NO measurement
The levels of NO in plasma and liver tissue were determined with NO assay kit according to the manufacturer’s instructions. The values of NO were assessed according to the absorbance measured at 540 nm and the hepatic levels of NO were normalized by the total protein concentration of each sample.
MPO activity assay
The frozen liver tissues were homogenized in phosphate buffer containing 0.5% hexadecyltrimethylammonium bromide. The activities of MPO were determined with MPO assay kit according to the manufacturer’s instructions. The values of MPO were assessed according to the absorbance measured at 450 nm and normalized by the total protein concentration of each sample.
Caspase activities determination
The hepatic activities of caspase-3, caspase-8 and caspase-9 were determined using colorimetric assay kits according to the manufacturer’s instructions. Briefly, the liver samples were homogenized in cell lysis buffer, the homogenates were centrifuged for 1 min at 10,000 g and the supernatant was incubated with Ac-DEVD-pNA, Ac-IETD-pNA and Ac-LEHD-pNA (substrates for caspase-3, caspase-8 and caspase-9, respectively) for 90 min at 37℃. The activities of caspases were assessed according to the absorbance measured at 405 nm and normalized by the total protein concentration of the same sample.
The apoptotic hepatocytes were detected with an In Situ Cell Death Detection Kit (Roche) according to the manufacturer’s instructions. The terminal transferase reactions finally produced a dark-brown precipitate; then, the sections were counterstained slightly with hematoxylin.
Quantitative real-time PCR
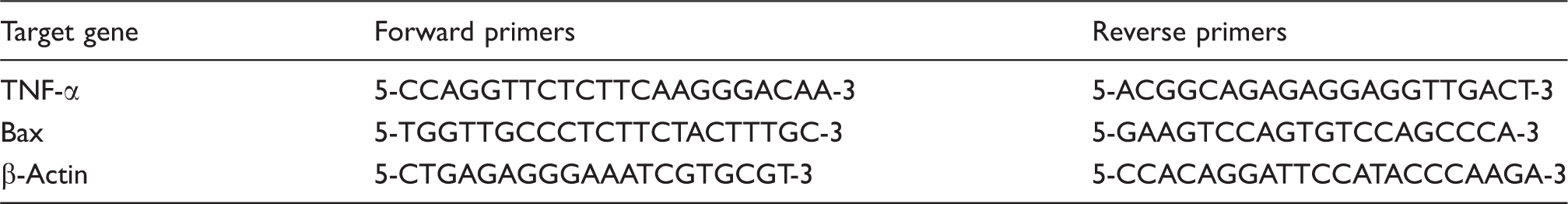
The mRNA level of TNF-α and Bax in liver were determined by quantitative real-time PCR. Briefly, total RNA was isolated from liver samples using Trizol reagent. First-strand cDNA was synthesized using oligo-dT primer and the Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase. Quantitative PCR was performed with SYBR green PCR Master Mix (Qiagen, Hilden, Germany) with the following conditions: denaturation at 95℃ for 10 s, annealing at 58℃ for 20 s and elongation at 72℃ for 20 s. The mRNA levels of TNF-α, erythropoietin and Bax were normalized by that of β-actin. The primers used to in the present studies are listed in Table 1.
Sequences of the primers used for quantitative real-time PCR.
Target gene
Forward primers
Reverse primers
TNF-α
5-CCAGGTTCTCTTCAAGGGACAA-3
5-ACGGCAGAGAGGAGGTTGACT-3
Bax
5-TGGTTGCCCTCTTCTACTTTGC-3
5-GAAGTCCAGTGTCCAGCCCA-3
β-Actin
5-CTGAGAGGGAAATCGTGCGT-3
5-CCACAGGATTCCATACCCAAGA-3
Western blot analysis
Total proteins from frozen liver samples were prepared according to the method described by the protein extract kit. The total protein concentration was determined using the BCA protein assay kit. Protein extracts were fractionated on 10% polyacrylamide SDS gel and then transferred to nitrocellulose membrane. The membrane was blocked with 5% (w/v) non-fat milk in Tris-buffered saline containing 0.05% Tween-20, and then the membrane was incubated with primary Ab overnight at 4℃, followed by incubation with secondary Ab. Ab binding was visualized with an ECL chemiluminescence system and short exposure of the membrane to X-ray films.
Statistical analysis
All data from the experiments were expressed as a mean ± SD. Statistical significance was determined by the Student's t-test for comparisons of two groups. Multigroup comparisons were performed using one-way ANOVA multiple comparisons among means, with the Turkey's post hoc test. Results were considered statistically significant when P < 0.05.
Results
AICAR attenuated LPS/D-Gal-induced liver injury
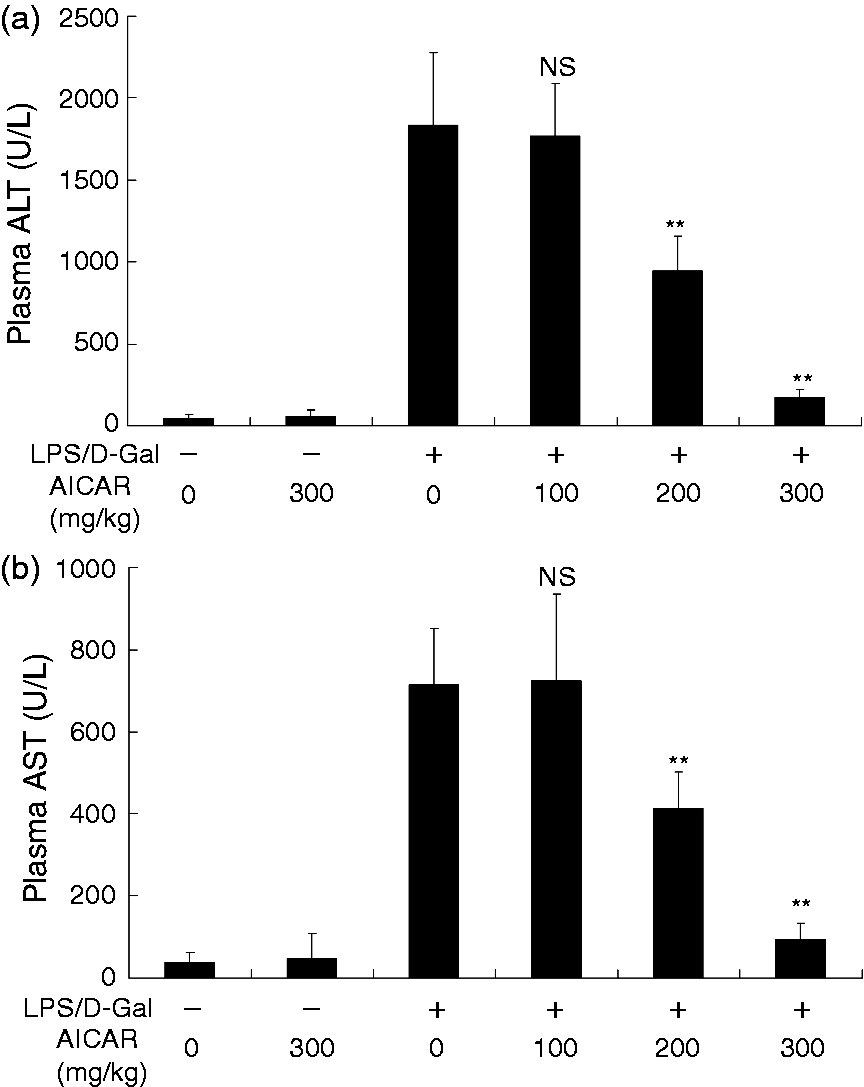
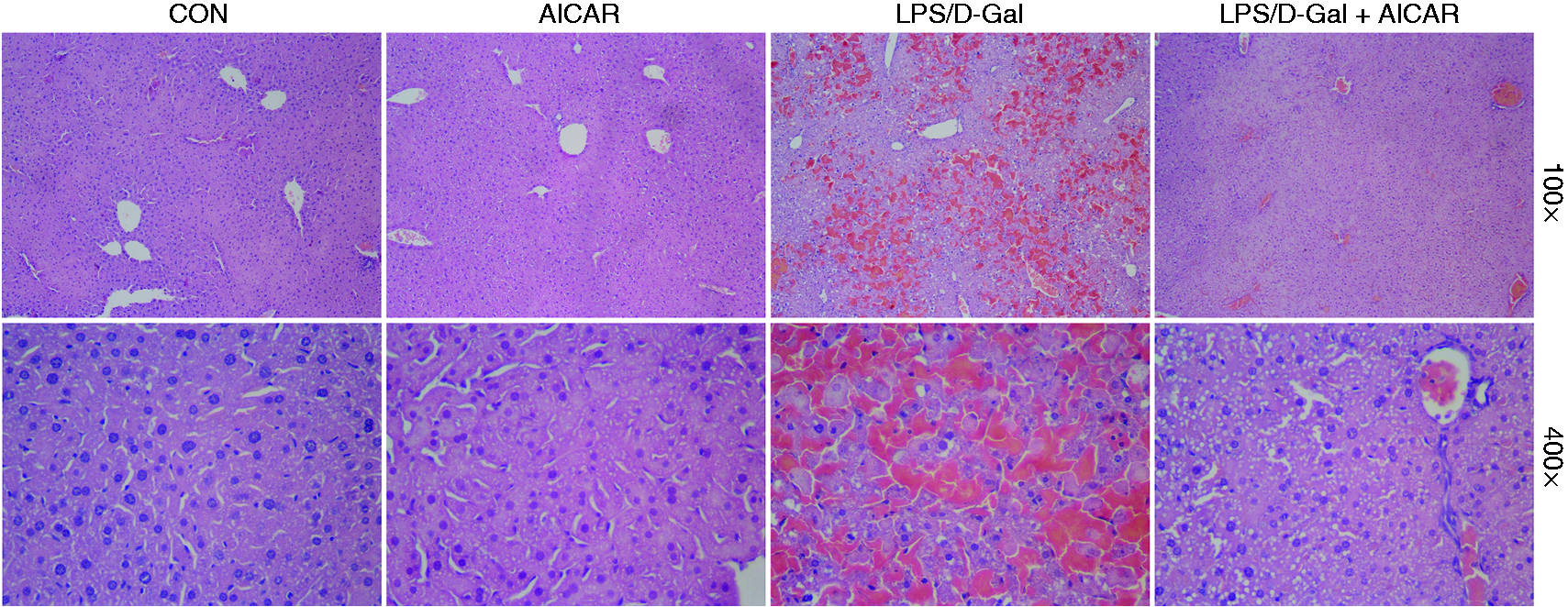
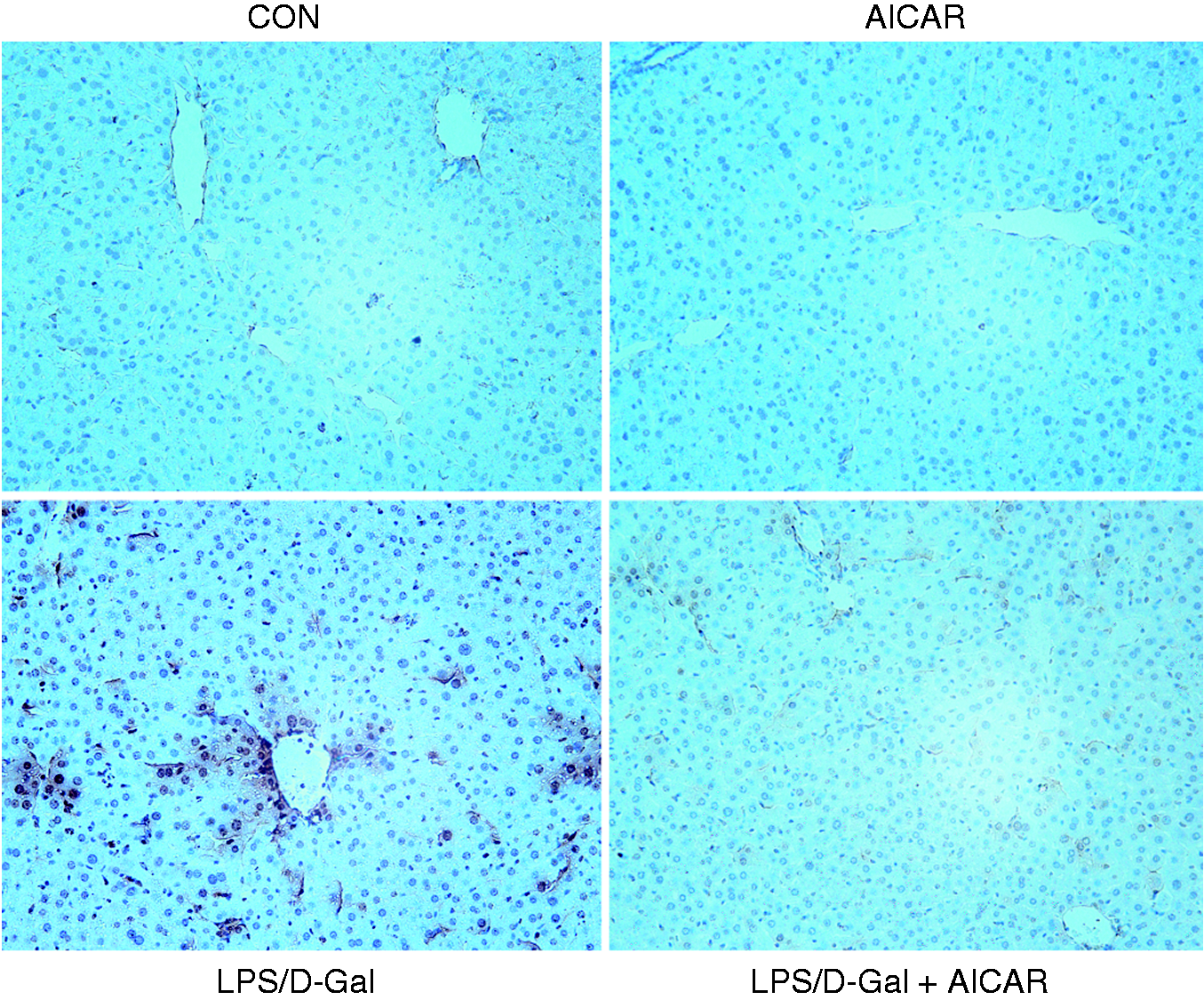
The plasma ALT and AST increased markedly in mice challenged with LPS/D-Gal but the elevation of ALT and AST was dose-dependently suppressed by AICAR (Figure 1a, b). Consistently, the degree of histological abnormalities, including severe congestion and destruction of hepatic architecture, in LPS/D-Gal-exposed mice were obviously alleviated in the AICAR-treated group (Figure 2). These data indicated that LPS/D-Gal-induced liver damage was attenuated by AICAR.
AICAR suppressed LPS/D-Gal-induced elevation of plasma aminotransferases. Mice were treated with vehicle or AICAR (100 mg/kg, 200 mg/kg and 300 mg/kg) in the presence or absence of LPS/D-Gal exposure. The plasma levels of (a) ALT and (b) AST were determined at 6 h after LPS/D-Gal exposure. Data are expressed as mean ± SD, n = 8. NSP > 0.05, **P < 0.01, compared with the LPS/D-Gal group (LPS/D-Gal +/AICAR 0).
AICAR improved LPS/D-Gal-induced histological abnormalities. Mice were treated with vehicle or AICAR (300 mg/kg) in the presence or absence of LPS/D-Gal exposure. Liver samples were harvested at 6 h after LPS/D-Gal exposure. Liver sections were stained with hematoxylin and eosin for morphological evaluation and the representative liver sections of each group are shown. CON: control.
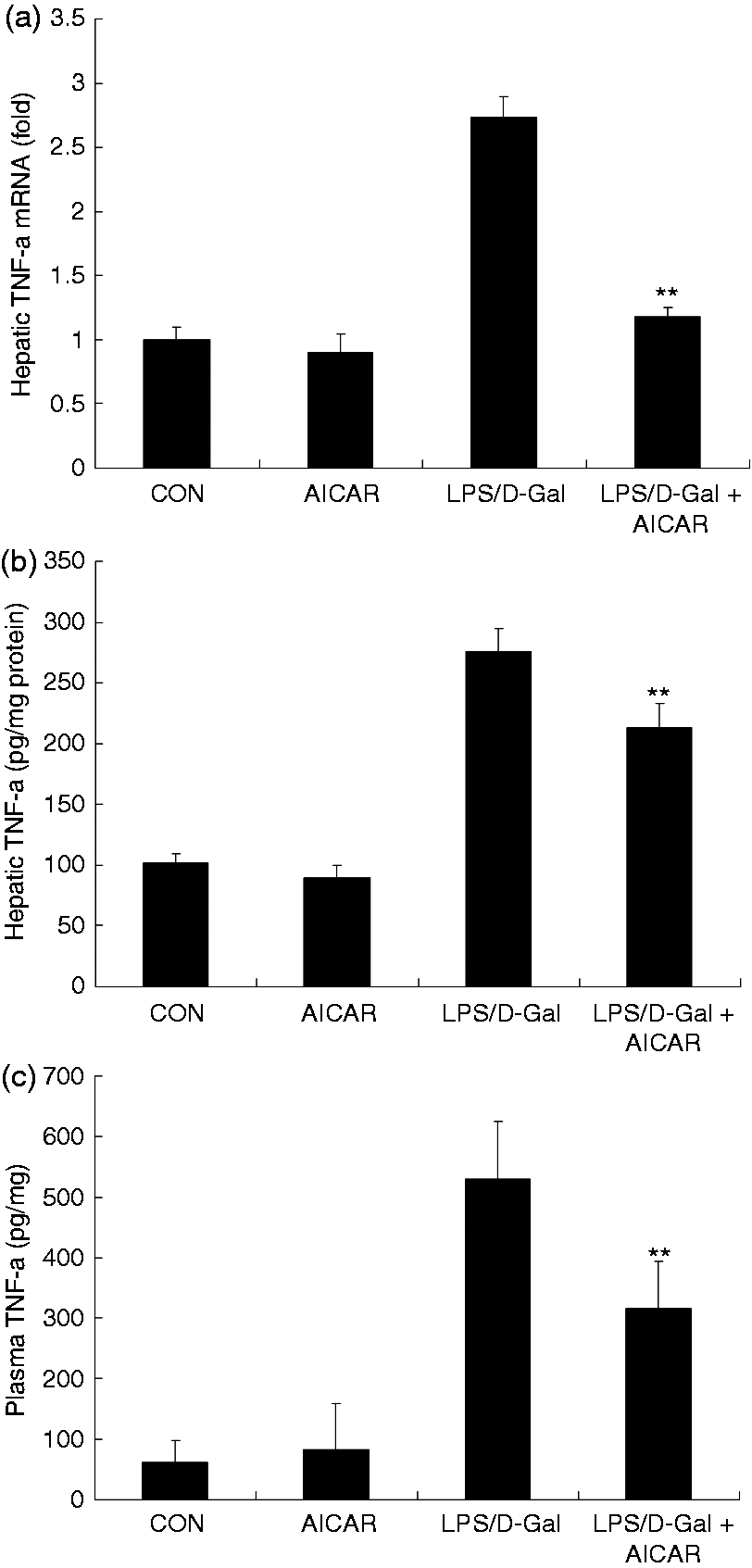
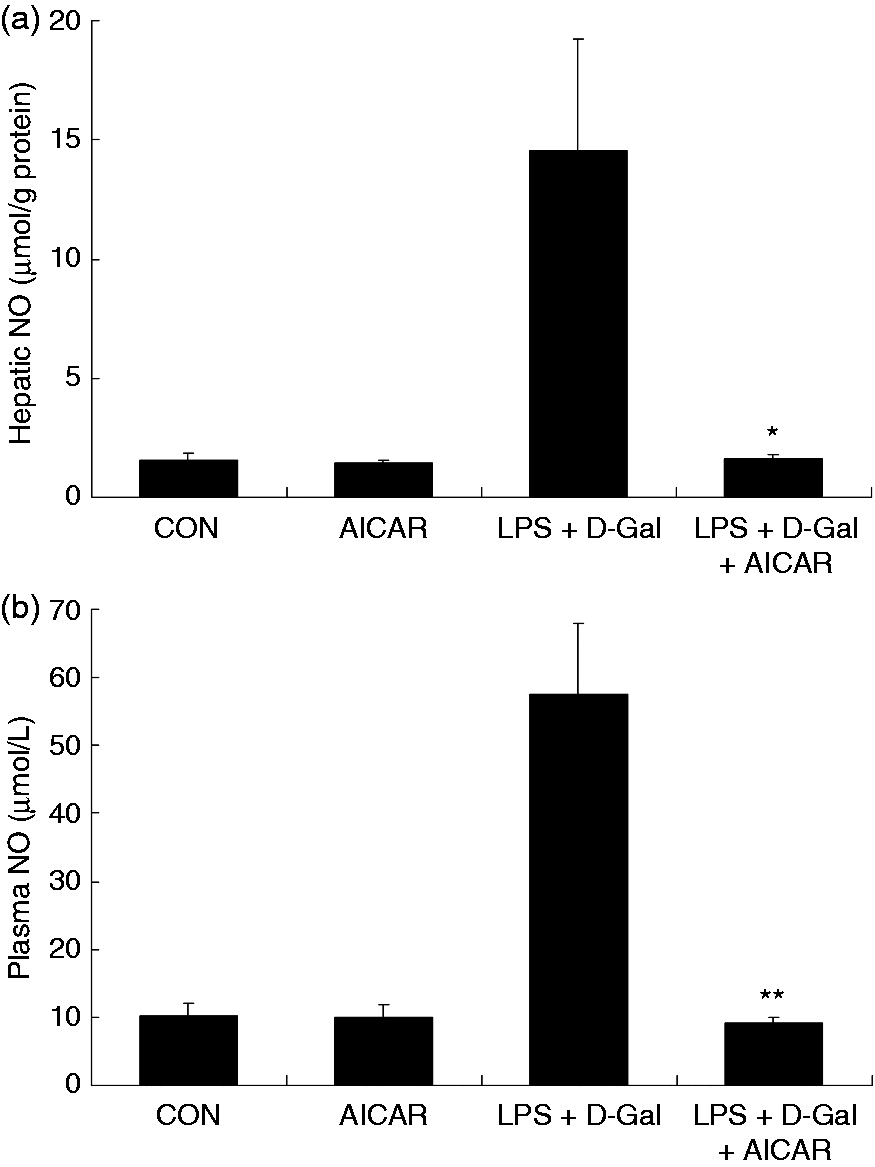
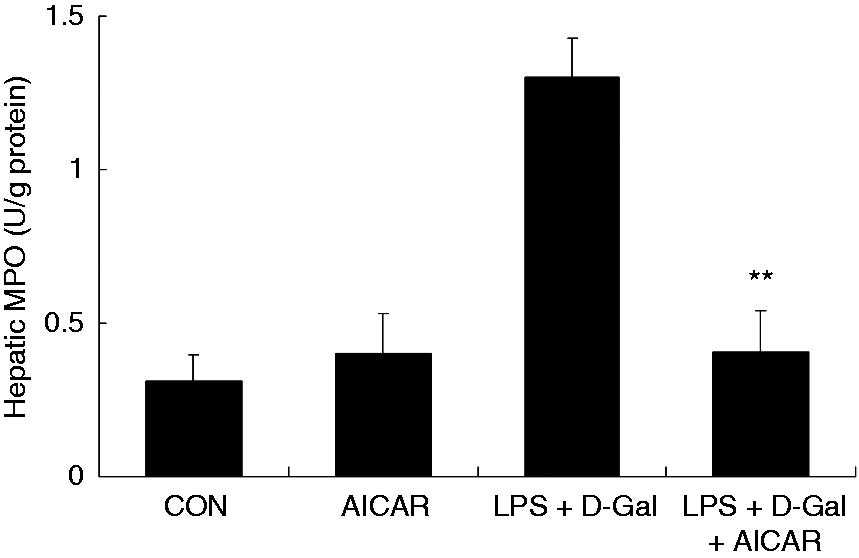
Challenge with LPS/D-Gal induced the up-regulation of TNF-α in liver tissue and plasma, whereas treatment with AICAR significantly reduced the level of TNF-α (Figure 3a–c). In parallel, the elevation of NO and MPO induced by LPS/D-Gal was also suppressed by AICAR (Figures 4 and 5). These data indicated that LPS/D-Gal-induced inflammatory response was suppressed by AICAR.
AICAR suppressed LPS/D-Gal-induced expression of TNF-α. Mice were treated with vehicle or AICAR (300 mg/kg) in the presence or absence of LPS/D-Gal exposure. (a) The mRNA levels of TNF-α in liver, (b) the protein levels of TNF-α in plasma and (c) liver were determined. Data are expressed as mean ± SD, n = 8. **P < 0.01 compared with the LPS/D-Gal group. CON: control.
AICAR suppressed LPS/D-Gal-induced production of NO. Mice were treated with vehicle or AICAR (300 mg/kg) in the presence or absence of LPS/D-Gal exposure. The levels of NO in (a) plasma and (b) liver were determined. Data are expressed as mean ± SD, n = 8. *P < 0.05, **P < 0.01, compared with the LPS/D-Gal group. CON: control.
AICAR suppressed LPS/D-Gal-induced up-regulation of MPO. Mice were treated with vehicle or AICAR (300 mg/kg) in the presence or absence of LPS/D-Gal exposure. The hepatic activities of MPO were determined. Data are expressed as mean ± SD, n = 8. **P < 0.01, compared with the LPS/D-Gal group. CON: control.
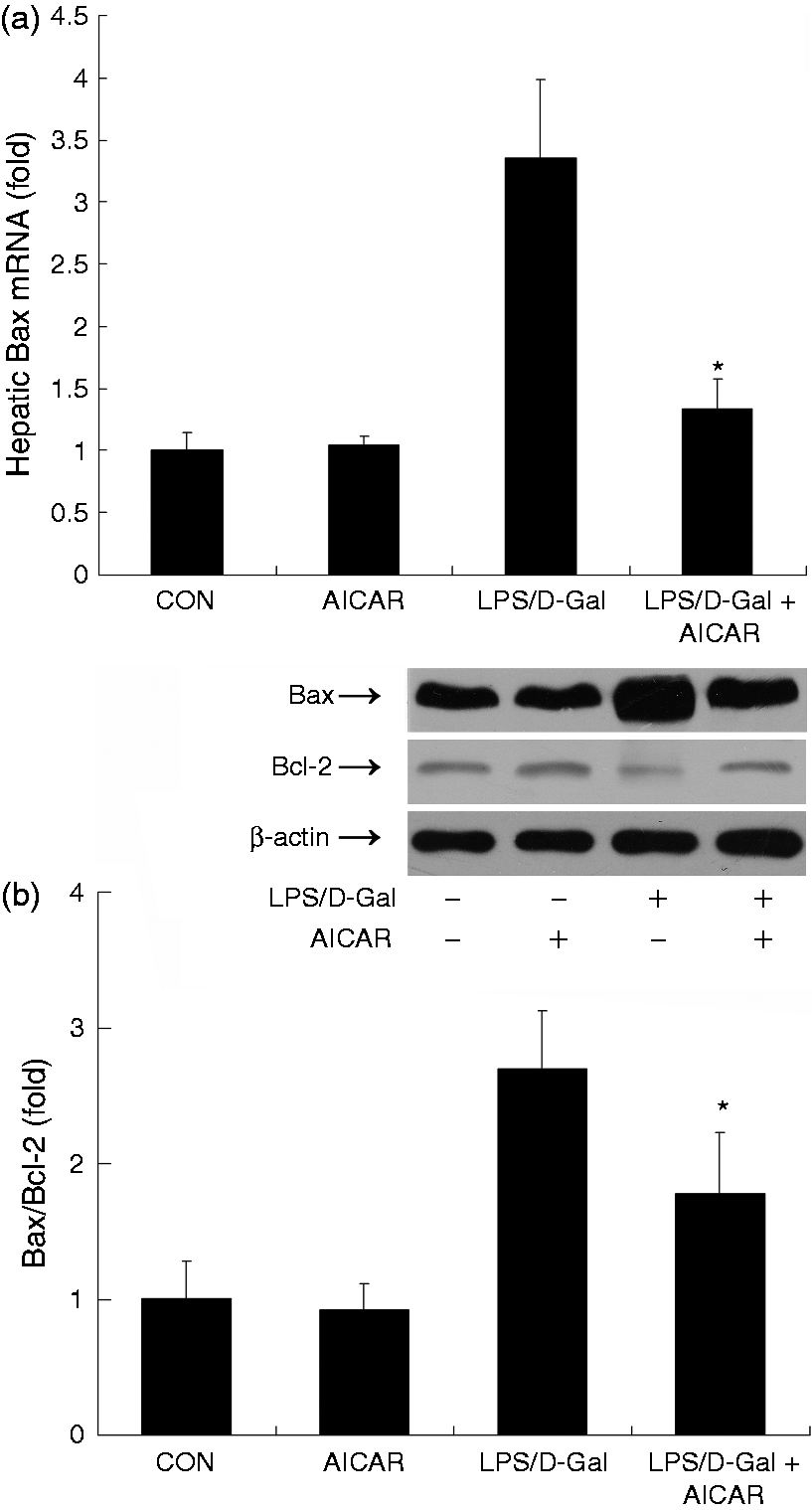
AICAR decreased LPS/D-Gal-induced up-regulation of Bax/Bcl-2 ratio
Apoptosis is one of the typical features of LPS/D-Gal-induced liver damage.3 The mRNA and protein levels of Bax, a representative pro-apoptotic factor,18 increased in mice exposed to LPS/D-Gal, while AICAR significantly suppressed the induction of Bax and reduced the elevation of Bax/Bcl-2 ratio (Figure 6).
AICAR suppressed LPS/D-Gal-induced up-regulation of Bax. Mice were treated with vehicle or AICAR (300 mg/kg) in the presence or absence of LPS/D-Gal exposure. Liver samples were harvested at 6 h after LPS/D-Gal exposure. (a) The mRNA levels of Bax and (b) the protein levels of Bax and Bcl-2 were determined. The bands of Bax, Bcl-2 and β-actin are indicated by arrows, and the blots were scanned by densitometry and data presented as relative intensity units. Data are expressed as mean ± SD, n = 8. *P < 0.05 compared with the LPS/D-Gal group. CON: control.
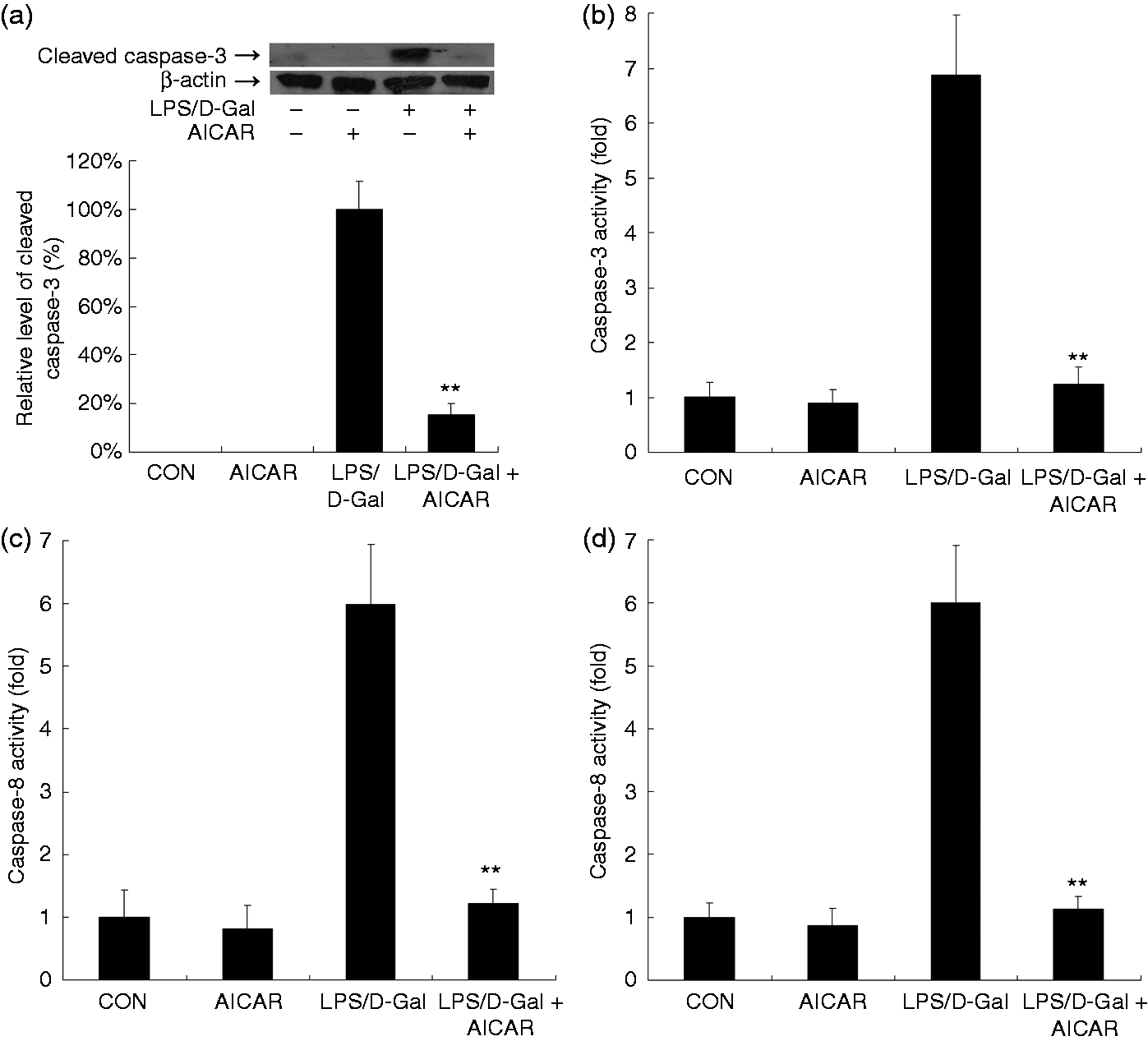
AICAR suppressed LPS/D-Gal-induced apoptosis
The immunoblot analysis indicated that AICAR suppressed the LPS/D-Gal-induced up-regulation of cleaved caspase-3 (Figure 7a). Consistently, treatment with AICAR inhibited the increased caspase-3, caspase-8 and caspase-9 activities in LPS/D-Gal-exposed mice (Figure 7b–d). The TUNEL assay indicated that massive TUNEL-positive apoptotic hepatocytes were presented in the liver of LPS/D-Gal-exposed mice but the count of TUNEL-positive cells decreased after AICAR treatment (Figure 8). These data indicated that LPS/D-Gal-induced hepatocyte apoptosis was suppressed by AICAR.
AICAR suppressed LPS/D-Gal-induced up-regulation of cleaved caspase-3 and caspase activities. Mice were treated with vehicle or AICAR (300 mg/kg) in the presence or absence of LPS/D-Gal exposure. Liver samples were harvested at 6 h after LPS/D-Gal exposure. (a) The levels of cleaved caspase-3 were determined by immunoblot analysis. The bands of cleaved caspase-3 and β-actin are indicated by arrows, and the blots were scanned by densitometry and data presented as relative intensity units. In addition, the activities of (b) caspase-3, (c) caspase-8 and (d) caspase-9 were determined. Data are expressed as mean ± SD, n = 8. **P < 0.01 compared with the LPS/D-Gal group. CON: control.
AICAR suppressed LPS/D-Gal-induced apoptosis. Mice were treated with vehicle or AICAR (300 mg/kg) in the presence or absence of LPS/D-Gal exposure. Liver samples were harvested at 6 h after LPS/D-Gal exposure. The apoptotic cells were determined by TUNEL assay and the TUNEL-positive cells showed a dark-brown nucleus. Representative liver sections of each group are shown (original magnification 200×). CON: control.
Discussion
AMPK is a pivotal energy sensor that engages in the maintenance of energy homeostasis and involves in the regulation of diverse energy-intensive biological processed such as proliferation, apoptosis and inflammation.5,7,14 AICAR is widely used as a pharmacological activator of AMPK for the investigation of the physiological functions and the pathophysiological significances of AMPK.15,16 In the present study, we found that AICAR significantly attenuated LPS/D-Gal-induced acute liver injury. The protective benefits were evidenced by suppressed elevation of plasma aminotransferases and improved histopathological abnormality.
The liver injury induced by LPS/D-Gal largely depends on the activation of inflammatory cells and the production of inflammatory mediators.3 In the present study, the LPS/D-Gal-induced up-regulation of the representative inflammatory mediators, including TNF-α and NO, and the inflammatory biomarker MPO was significantly suppressed by AICAR, suggesting that AICAR might have anti-inflammatory effects in LPS/D-Gal-exposed mice. In agreement with our findings, the anti-inflammatory effects of AICAR have been confirmed under various inflammatory circumstances. It was reported that AICAR suppressed LPS-induced expression of TNF-α and cyclooxygenase-2 in macrophages.19,20 AICAR also inhibited TNF-α production induced by free fatty acids, and long-term administration of low-dose AICAR significantly suppressed adipose inflammation in diet-induced obese mice.10,21 In addition, treatment with AICAR alleviated inflammatory lung injury insulted by lipoteichoic acid, LPS, toluene polyinosinic-polycytidylic acid [poly (I:C)] or diisocyanate, and protected against colitis induced by dextran sulfate sodium or 2,4,6-trinitrobenzene sulfonic acid.16,17,22–25
Apoptosis constitutes a hallmark in LPS/D-Gal-induced liver damage.3 In the present study, LPS/D-Gal-induced elevation in cleaved caspase-3, caspases activities and TUNEL-positive hepatocytes was significantly inhibited by AICAR, indicating that AICAR suppressed hepatocyte apoptosis in LPS/D-Gal-exposed mice. In the principle of LPS/D-Gal-induced liver damage, TNF-α is a major deleterious factor with potent pro-apoptotic activity,3 while the TNF-α receptor 1-deficient mice are resistant to LPS/D-Gal-induced liver damage.26 Therefore, the anti-apoptotic effects of AICAR might result from its inhibitory effects on TNF-α production.
In addition to the inflammatory suppressive effects, increasing evidence suggests that AMPK activation might be involved in apoptosis regulation. It has been reported that incubation with the AMPK activator AICAR or expression of a constitutively active AMPK suppressed hyperglycemia-induced apoptosis in human umbilical vein endothelial cells.27 AMPK could also protect neurons from Glc deprivation-induced apoptosis because suppression of AMPK using antisense technology resulted in enhanced neuronal death following Glc deprivation and the neuroprotective effect of AICAR was abolished by AMPK suppression.28 This evidence suggested that AMPK might have an anti-apoptotic function; however, there have also been controversial experimental results. For example, AMPK contributed to UV-induced apoptosis in human skin keratinocytes because AMPK-specific small interfering RNA knockdown inhibited UV-induced apoptosis.29 Activation of AMPK by AICAR or adenoviral expression of constitutively active AMPK could also induce apoptosis in the beta-cell line.30 Therefore, AMPK might have diverse and context-dependent regulatory effects on apoptosis. The direct roles of AMPK in LPS/D-Gal-induced apoptosis remain to be further determined.
Taken together, the present study has shown that the AMPK activator AICAR effectively attenuated LPS/D-Gal-induced acute hepatitis, and that these effects were associated with reduced production of pro-inflammatory mediators. Therefore, the protective benefits of AICAR in LPS/D-Gal-induced acute hepatitis might be associated with its anti-inflammatory effects. Although the exact role of AMPK in liver disorders induced by LPS/D-Gal or other pathological factors remain, the present experimental data suggest that AMPK might become a novel target for the treatment of inflammation-based liver disorders.
Footnotes
Funding
This work was supported by grants from the National Nature Science Foundation of China (No. 81370179) and Chongqing Municipal Education Commission (No. KJ1400235).
Conflict of interest
The authors do not have any potential conflicts of interest to declare.
References
1.
YethonJAWhitfieldC. Lipopolysaccharide as a target for the development of novel therapeutics in gram-negative bacteria. Curr Drug Targets Infect Disord2001; 1: 91–106.
2.
TrentMSSteadCMTranAXHankinsJV. Diversity of endotoxin and its impact on pathogenesis. J Endotoxin Res2006; 12: 205–223.
TunonMJAlvarezMCulebrasJMGonzalez-GallegoJ. An overview of animal models for investigating the pathogenesis and therapeutic strategies in acute hepatic failure. World J Gastroenterol2009; 15: 3086–3098.
5.
HardieDGRossFAHawleySA. AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol2012; 19: 1222–1236.
6.
CarlingDThorntonCWoodsASandersMJ. AMP-activated protein kinase: new regulation, new roles?Biochem J2012; 445: 11–27.
7.
DunlopEATeeAR. The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem Soc Trans2013; 41: 939–943.
8.
RudermanNBCarlingDPrentkiMCacicedoJM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest2013; 123: 2764–2772.
9.
PearceELPoffenbergerMCChangCHJonesRG. Fueling immunity: insights into metAbolism and lymphocyte function. Science2013; 342: 1242454–1242454.
10.
YangZKahnBBShiHXueBZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem2010; 285: 19051–19059.
11.
GiriSNathNSmithB. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci2004; 24: 479–487.
12.
TsalikisJCroitoruDOPhilpottDJGirardinSE. Nutrient sensing and metabolic stress pathways in innate immunity. Cell Microbiol2013; 15: 1632–1641.
13.
O'NeillLAHardieDG. MetAbolism of inflammation limited by AMPK and pseudo-starvation. Nature2013; 493: 346–355.
14.
FerriN. AMP-activated protein kinase and the control of smooth muscle cell hyperproliferation in vascular disease. Vascul Pharmacol2012; 56: 9–13.
15.
WongAKHowieJPetrieJRLangCC. AMP-activated protein kinase pathway: a potential therapeutic target in cardiometAbolic disease. Clin Sci (Lond)2009; 116: 607–620.
16.
ZhaoXZmijewskiJWLorneE. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol2008; 295: L497–L504.
17.
BaiAMaAGYongM. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem Pharmacol2010; 80: 1708–1717.
18.
LAbiVErlacherMKiesslingSVillungerA. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ2006; 13: 1325–1338.
KuoCLHoFMChangMY. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J Cell Biochem2008; 103: 931–940.
21.
YangZWangXHeY. The full capacity of AICAR to reduce obesity-induced inflammation and insulin resistance requires myeloid SIRT1. PLoS One2012; 7: e49935–e49935.
22.
HoogendijkAJPinhancosSSvan der PollTWielandCW. AMP-activated protein kinase activation by 5-aminoimidazole-4-carbox-amide-1-beta-D-ribofuranoside (AICAR) reduces lipoteichoic acid-induced lung inflammation. J Biol Chem2013; 288: 7047–7052.
23.
ParkSJLeeKSKimSR. AMPK activation reduces vascular permeability and airway inflammation by regulating HIF/VEGFA pathway in a murine model of toluene diisocyanate-induced asthma. Inflamm Res2012; 61: 1069–1083.
24.
KimTBKimSYMoonKA. Five-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside attenuates poly (I:C)-induced airway inflammation in a murine model of asthma. Clin Exp Allergy2007; 37: 1709–1719.
25.
BaiAYongMMaAG. Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J Pharmacol Exp Ther2010; 333: 717–725.
26.
RotheJMackayFBluethmannH. Phenotypic analysis of TNFR1-deficient mice and characterization of TNFR1-deficient fibroblasts in vitro. Circ Shock1994; 44: 51–56.
27.
IdoYCarlingDRudermanN. Hyperglycemia-induced apoptosis in human umbilical vein endothelial cells: inhibition by the AMP-activated protein kinase activation. Diabetes2002; 51: 159–167.
28.
CulmseeCMonnigJKempBEMattsonMP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci2001; 17: 45–58.
29.
CaoCLuSKivlinR. AMP-activated protein kinase contributes to UV- and H2O2-induced apoptosis in human skin keratinocytes. J Biol Chem2008; 283: 28897–28908.
30.
KefasBACaiYLingZ. AMP-activated protein kinase can induce apoptosis of insulin-producing MIN6 cells through stimulation of c-Jun-N-terminal kinase. J Mol Endocrinol2003; 30: 151–161.