
Abstract
The renin–angiotensin system is classically regarded as a crucial regulator of circulatory homeostasis, but recent studies also revealed its pro-inflammatory roles. The beneficial effects of the angiotensin-converting enzyme inhibitor (ACEI) in severe inflammatory injury in the lung and heart have been previously reported, but its potential effects on lethal hepatitis were unknown. In this study, a mouse model with LPS/d-galactosamine (GalN)-induced fulminant hepatitis were used to test the protective potential of captopril, a representative ACEI. The results indicated that treatment with captopril significantly decreased the plasma level of alanine aminotransferase and aspartate aminotransferase, alleviated the histopathological damage of the liver tissue and improve the survival rate of LPS/GalN-challenged mice. These effects were accompanied by reduced mRNA levels of TNF-α and IL-6 in the liver, and decreased protein level of TNF-α and IL-6 in the plasma. In addition, the activation of caspases 3, 8 and 9, and the presence of TUNEL-positive apoptotic cells, were also suppressed by captopril treatment. The above evidence suggested that the renin–angiotensin system might be involved in the development of LPS/GalN-induced fulminant hepatitis and ACEI might have potential value in lethal hepatitis.
Introduction
The renin–angiotensin system (RAS) is a classic endocrine hormonal system that plays critical roles in the regulation of vascular resistance and the maintenance of fluid balance. 1 Angiotensin-converting enzyme (ACE) is the key enzyme catalyzing the conversion of angiotensin I to angiotensin II, which has become a valuable pharmacological target for blocking the formation of the downstream effectors and inhibiting the deleterious effects of RAS. 2 ACE inhibitors (ACEI) are currently used as a first-line antihypertensive drugs worldwide. 3
In addition to its central roles in the regulation of circulatory homeostasis, more extensive physiological and pathophysiological effects of RAS have been revealed recently. 4 For example, RAS is involved in the progress of inflammation-based tissue injury and angiotensin II is emerging as a powerful pro-inflammatory mediator.5,6 Therefore, ACEI might have potential value in preventing inflammatory injury, and the therapeutic benefits of ACEI have been confirmed in experimental animals with acute lung injury,7,8 myocarditis, 9 colitis10,11 and glomerulonephritis. 12
Because of its unique blood-supply pathway and histological characters, the liver is another vital organ susceptible to severe inflammatory injury. 13 Although the anti-inflammatory effects of ACEI have been observed in inflammation-based chronic hepatic disorders such as cirrhosis and steatohepatitis,14,15 the pharmacological significance of ACEI in life-threatening fulminant hepatitis remains unknown. In the present study, the potential effects of captopril, a representative ACEI, 16 were investigated in a mouse model with LPS/d-galactosamine (GalN)-induced lethal fulminant hepatitis.
Materials and methods
Materials
The chemical reagents, including captopril, LPS (from Escherichia coli, 055:B5) and GalN, were from Sigma (St. Louis, MO, USA). The alanine aminotransferase (ALT) assay kit and the aspartate aminotransferase (AST) assay kit were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). The ELISA kits for detecting mouse TNF-α and IL-6 were purchased from NeoBioscience Technology Company (Shenzhen, China). The total protein extract kit and the caspase-3, caspase-8 and caspase-9 colorimetric assay kits were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). An In Situ Cell Death Detection Kit was purchased from Roche (Indianapolis, IN, USA). The Abs for detecting cleaved caspase-3 and β-actin were from Cell Signaling Technology (Danvers, MA, USA). The BCA protein assay kit, the HRP-conjugated goat anti-rabbit Ab and the enhanced chemiluminescence (ECL) reagents were from Pierce Biotechnology (Rockford, IL, USA).
Animals
Male BALB/c mice weighing 20–25 g were provided by the Experimental Animal Center of Chongqing Medical University. All animals were fed with a standard laboratory diet and provided with water ad libitum. They were housed in a specific pathogen-free room at a temperature of 20–25℃ and 50 ± 5% relative humidity under a 12-h dark/light cycle and acclimatized for at least l wk before use. Experiments were conducted in accordance with the Animal Care and Use Committee of the Chongqing Medical University Guidelines for the Care and Use of Laboratory.
Experimental design
The mice were randomly allocated into four groups: (1) the control (CON) group mice received vehicle treatment; (2) the captopril group mice received captopril (25 mg/kg, dissolved in normal saline, i.p.) only (the dose of captopril was determined according to previous studies); 17 (3) the LPS/GalN group mice received LPS/GalN challenge; (4) the LPS/GalN + captopril group mice received captopril (25 mg/kg) 0.5 h prior to LPS/GalN challenge. Lethal hepatitis was induced in the latter two groups by i. p. injection of LPS (10 µg/kg) combined with GalN (700 mg/kg), dissolved in 0.9% normal saline. The mice were sacrificed 1.5 and 6 h after LPS/GalN challenge (eight mice per group at each time point), the blood and liver tissue samples were harvested for further analysis.
Survival rate analysis
The survival of mice (n = 20 per group) was monitored for at least 7 d after LPS/GalN challenge. The numbers of dead mice were counted at intervals of 6 h and the cumulative survival curve was depicted using the Kaplan–Meier method.
Histology analysis
Liver tissues were fixed in 10% buffered formalin and embedded in paraffin. The paraffin-embedded samples were sectioned (4 µm) and stained with hematoxylin and eosin for histopathological evaluation under a light microscope (Olympus, Tokyo, Japan).
Evaluation of plasma AST and ALT activities
The plasma activities of AST and ALT were assessed at 6 h after LPS/GalN exposure, using the commercial kits (Nanjing Jiancheng Biotech, Nanjing, China). The enzymatic activities of AST and ALT were expressed as units/liter (U/l).
Evaluation of plasma TNF-α and IL-6 level
The plasma TNF-α and IL-6 level were determined at 1.5 h following LPS/GalN treatment by using the commercially available ELISA kits (NeoBioscience), according the manufacturer's protocol.
Quantitative real-time PCR analysis
Total RNA was isolated from liver samples using Trizol reagent, according to the manufacture's instructions, and quantitative PCR was performed with SYBR green PCR Master Mix. The following primers were used to amplify TNF-α cDNA: sense, 5′-CGGGCAGGTCTACTTTGGAG-3′; antisense, 5′-CAGGTCACTGTCCCAGCATC-3′. The following primers were used to amplify IL-6 cDNA: sense, 5′-AGTTGCCTTCTTGGGACTGATG-3′; antisense, 5′-CTCATTTCCACGATTTCCCAG-3′. The following primers were used to amplify β-actin cDNA: sense, 5′-CTGAGAGGGAAATCGTGCGT-3′; antisense, 5′-CCACAGGATTCCATACCCAAGA-3′.
PCR was performed using the following PCR conditions: denaturing at 95℃ for 15 s, annealing at 58℃ for 15 s, and elongation at 72℃.
Detection of hepatocyte apoptosis
The assessment of liver apoptosis was performed with the In Situ Cell Death Detection Kit according to the manufacturer's instructions (Roche). The terminal transferase reactions finally produced a dark-brown precipitate and then the sections were counterstained slightly with hematoxylin. The numbers of TUNEL-positive cells in 10 randomly selected high-power fields (×400) were counted under microscopy
Determination of the hepatic caspase activity
The liver samples were homogenized in lysis buffer. After centrifugation for 15 min at 25,000 g, the supernatant was incubated with a substrate peptide [Ac-DEVD-pNA for caspase-3, Ac-IETD-pNA for caspase-8 and Ac-LEHD-pNA for caspase-9 (Beyotime)] for 90 min at 37℃. The activities of caspases were assessed according the absorbance measured at 405 nm and normalized by the total protein concentration of the same sample.
Western blot analysis of cleaved caspase-3
Total proteins from frozen liver samples were prepared according to the method described by the protein extraction kit (Beyotime). The total protein concentration was determined using the BCA protein assay kit (Pierce). Protein extracts were fractionated on 10% polyacrylamide–SDS gel and then transferred to nitrocellulose membrane. The membrane was blocked with 5% (w/v) nonfat milk in Tris-buffered saline containing 0.05% Tween-20, and then the membrane was incubated with primary Ab overnight at 4℃, followed by incubation with the secondary Ab. Ab binding was visualized with an ECL chemiluminescence system and short exposure of the membrane to X-ray films (Kodak, Tokyo, Japan).
Statistical analysis
All results, except survival from the experiments, are presented as mean ± SD. Differences among multiple groups were compared by one-way ANOVA, followed by Tukey–Kramer’s multiple comparison test. The SPSS software (version 19; IBM, Armonk, NY, USA) was used to perform the statistical analysis. The level of significance was fixed at P < 0.05, with respect to all statistical tests. Survival data were analyzed by Kaplan–Meier curves and the log-rank test.
Results
Captopril ameliorated LPS/GalN-induced liver damage
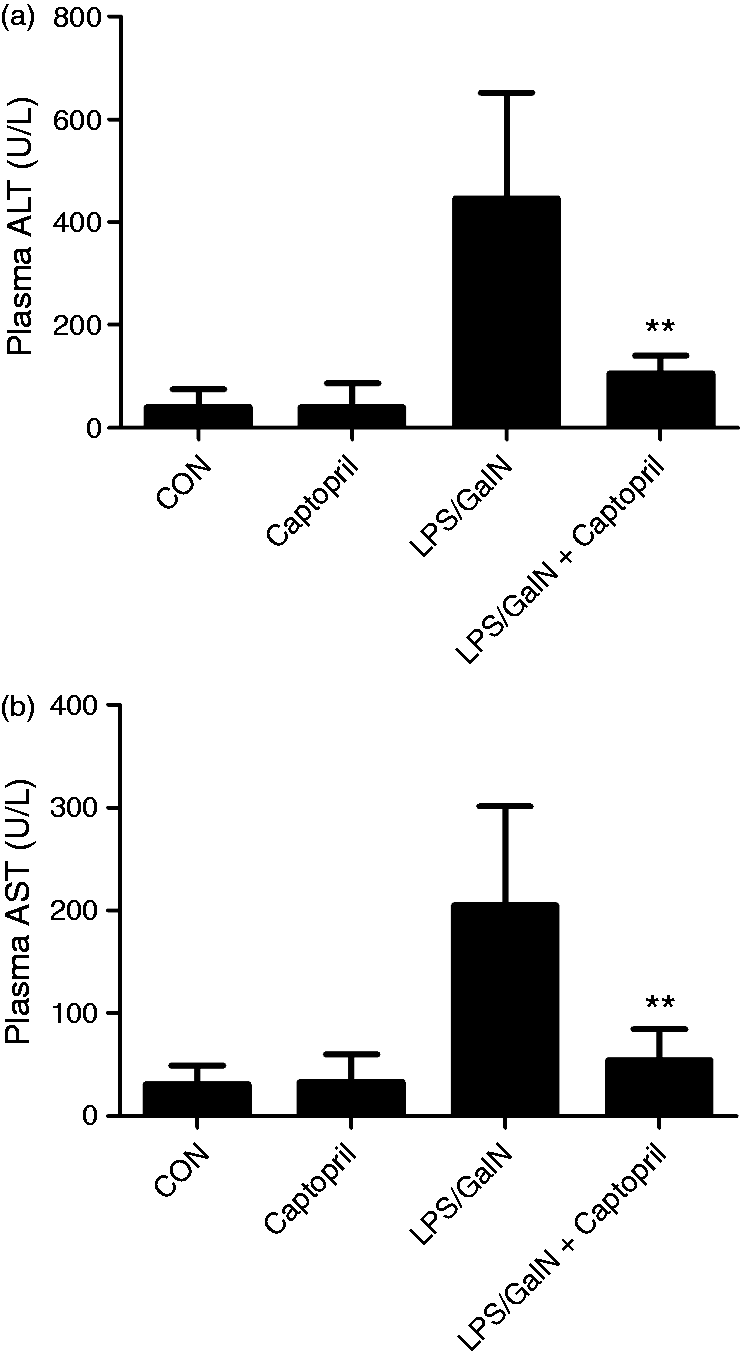
The histological features shown in Figure 1 indicated that the livers showed extensive areas with portal inflammation and hemorrhagic necrosis at 6 h after exposure to LPS/GalN. These pathological alterations were ameliorated in mice that received captopril treatment. In addition, the plasma activities of ALT and AST were significantly increased in the LPS/GalN group compared with the CON group, while treatment with captopril significantly attenuated these alteration (Figure 2).
Captopril ameliorated LPS/GalN-induced histological abnormalities. Mice were treated with vehicle or captopril in the absence or presence of LPS/GalN challenge and the liver samples were harvested at 6 h after LPS/GalN exposure. Histopathology of the representative liver sections of each group was shown (stained with hematoxylin and eosin, original magnification 100× and 400×). Captopril suppressed LPS/GalN-induced elevation of aminotransferases. Mice were treated with vehicle or captopril in the absence or presence of LPS/GalN challenge. The levels of (a) ALT and (b) AST in plasma were determined at 6 h after LPS/GalN exposure (n = 8; **P < 0.01, compared with the LPS/GalN group).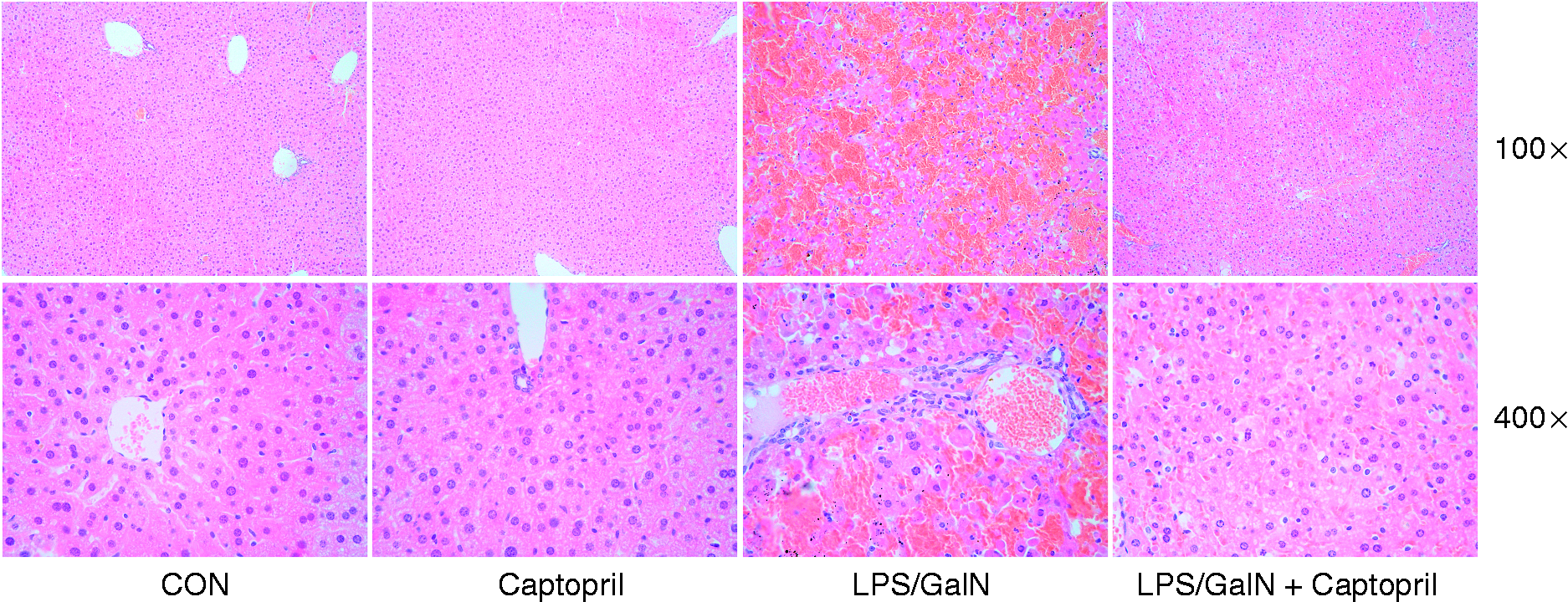
Captopril suppressed the expression of TNF-α and IL-6
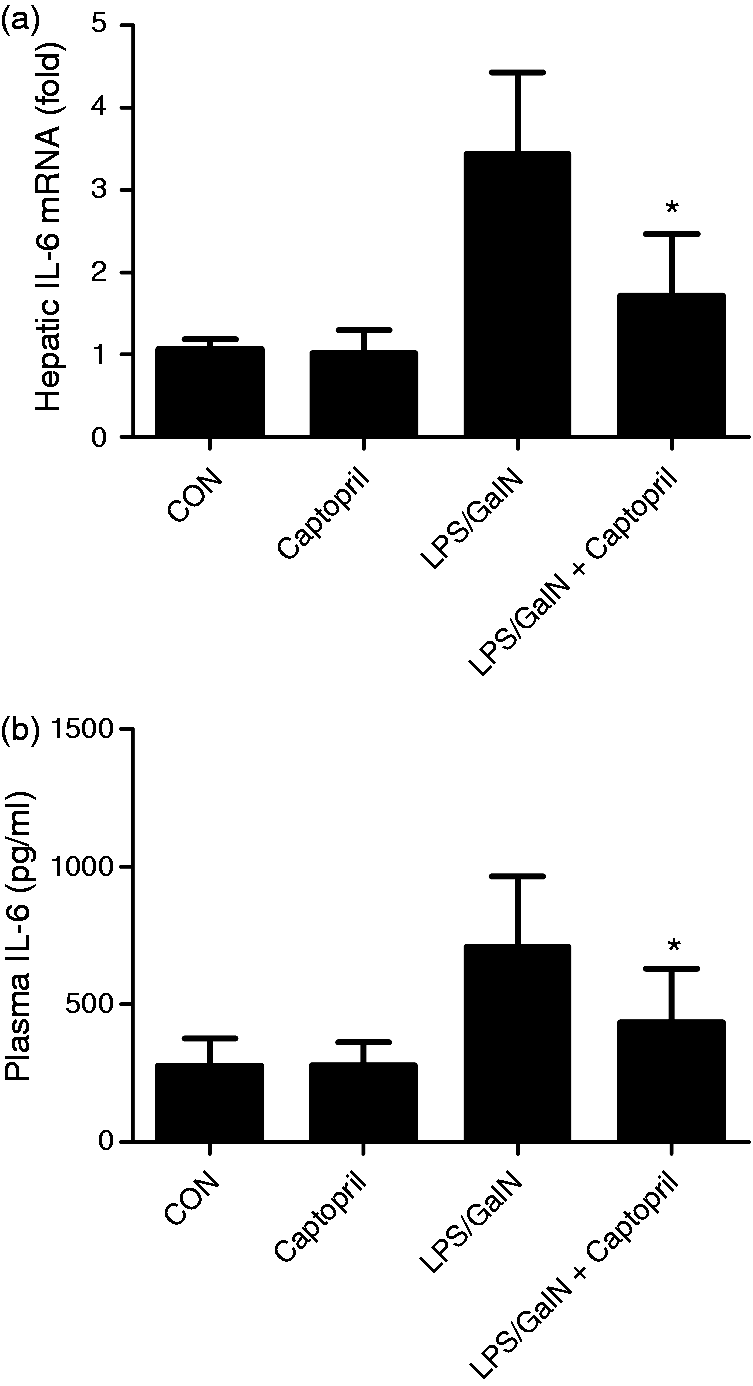
It is well known that TNF-α and IL-6 are crucial cytokines involved in the development of LPS/GalN-induced acute liver injury.18,19 In the present study, the mRNA level of TNF-α in liver and the protein level of TNF-α in plasma of LPS/GalN exposed mice were significantly suppressed by captopril (Figure 3). In agreement with these results, the mRNA and protein level IL-6 also decreased after captopril treatment (Figure 4).
Captopril inhibited LPS/GalN-induce TNF-α expression. Mice were treated with vehicle or captopril in the absence or presence of LPS/GalN challenge and the liver samples were harvested at 1.5 h after LPS/GalN exposure. (a) The mRNA level of TNF-α in liver was assessed by quantitative real-time PCR, the β-actin was used as an internal control. All values were normalized against β-actin and expressed as a proportion of the control. (b) The protein level of TNF-α in plasma was measured using ELISA kit (n = 8; *P < 0.05, **P < 0.01 compared with the LPS/GalN group). Captopril inhibited LPS/GalN-induced IL-6 expression. Mice were treated with vehicle or captopril in the absence or presence of LPS/GalN challenge and the liver samples were harvested at 1.5 h after LPS/GalN exposure. (a) The mRNA level of IL-6 in liver was assessed by quantitative real-time PCR, the β-actin was used as an internal control and all values were normalized against β-actin and expressed as a proportion of the control. (b) The protein level of IL-6 in plasma was measured using ELISA kit (n = 8; *P < 0.05, compared with the LPS/GalN group).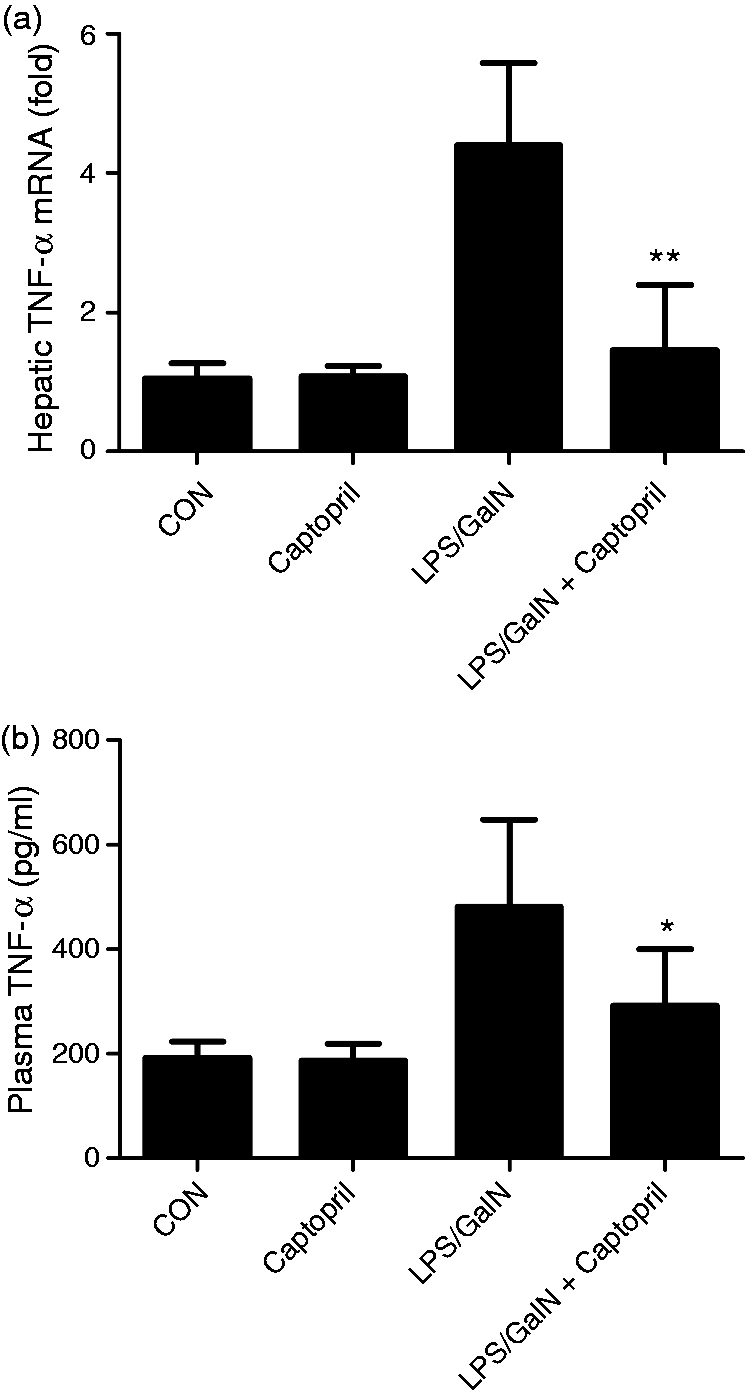
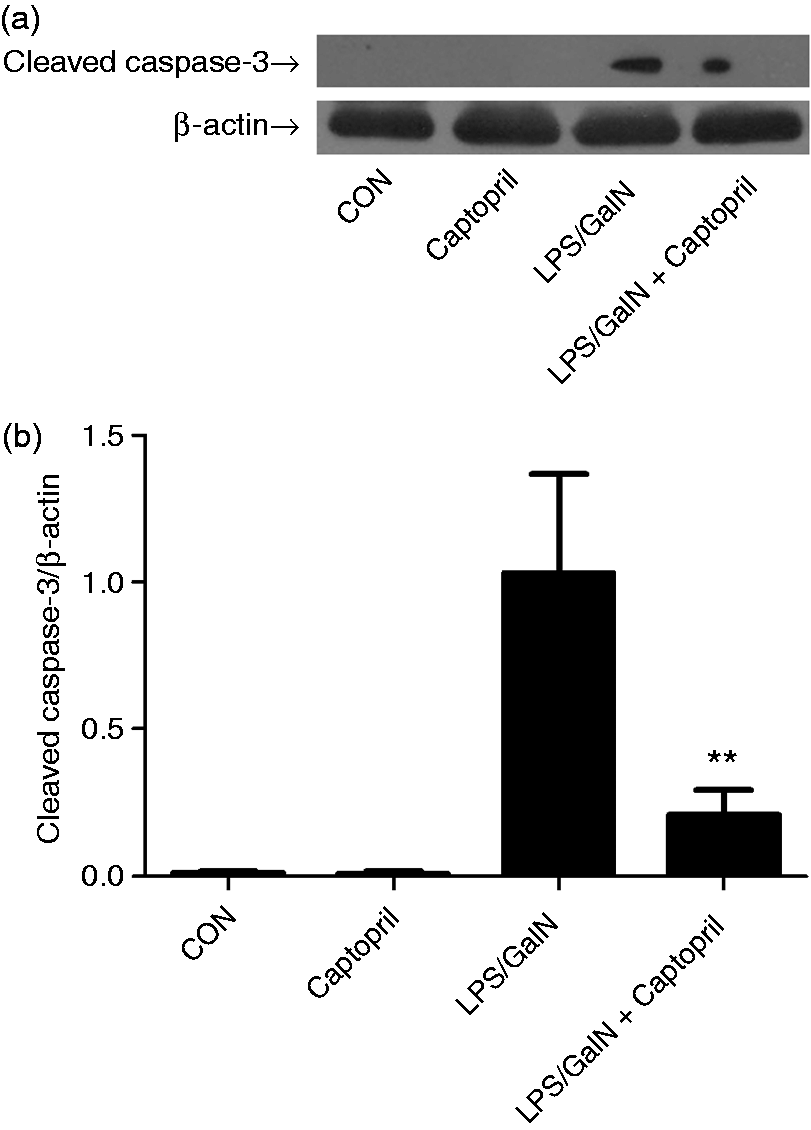
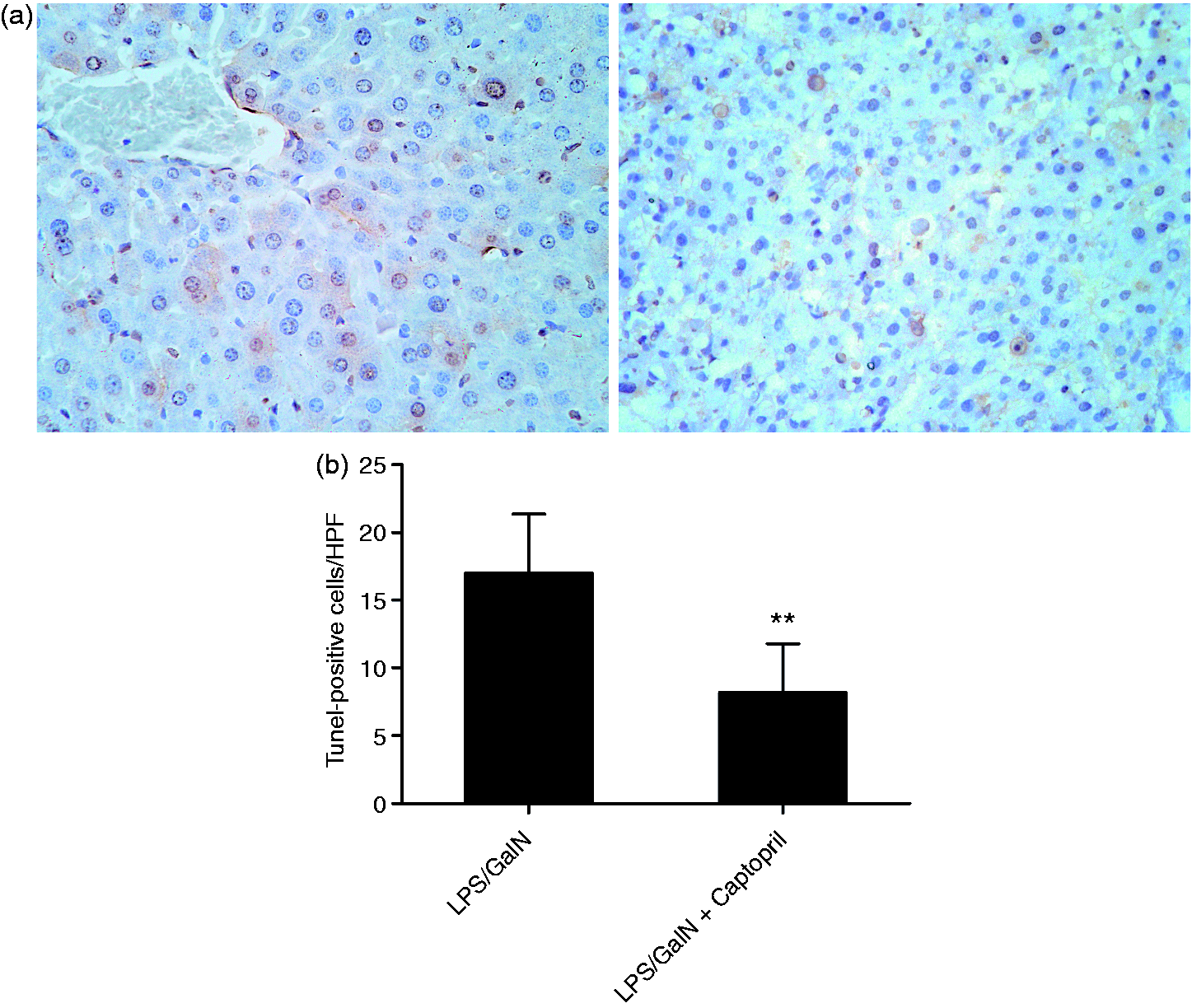
Captopril suppressed LPS/GalN-induced hepatocyte apoptosis
LPS/GalN exposure can induce massive apoptosis of hepatocytes.
20
The experiment suggested treatment with captopril inhibited the activation of apoptosis-related proteases caspase-3, caspase-8 and caspase-9 induced by LPS/GalN (Figure 5). Using immunoblot analysis, the increased level of cleaved caspase-3 in LPS/GalN-exposed mice was also down-regulated by captopril (Figure 6). Consistently, treatment with captopril remarkably decreased the number of TUNEL-positive cells as compared with the LPS/GalN group (Figure 7).
Captopril suppressed LPS/GalN-induced activation of caspases. Mice were treated with vehicle or captopril in the absence or presence of LPS/GalN challenge and the liver samples were harvested at 6 h after LPS/GalN exposure. The activities of (a) caspase-3, (b) caspase-8 and (c) caspase-9 in liver were determined (n = 8; *P < 0.05, compared with the LPS/GalN group). Captopril suppressed LPS/GalN-induced cleavage of caspase-3. Mice were treated with vehicle or captopril in the absence or presence of LPS/GalN challenge and the liver samples were harvested at 6 h after LPS/GalN exposure. (a) The level of cleaved caspase-3 was detected by Western blot analysis and β-actin was used as the internal control protein. (b) The blots were scanned and semi-quantified, the values were normalized against β-actin and expressed as a proportion of the control (n = 4; **P < 0.01, compared with the LPS/GalN group). Captopril suppressed LPS/GalN-induced hepatocyte apoptosis. (a) The apoptotic cells were determined by TUNEL assay and the representative liver sections of each group were shown (original magnification 400×). (b). The numbers of TUNEL-positive cells in 10 randomly selected high-power fields (400×) were counted under microscopy (n = 4; **P < 0.01, compared with the LPS/GalN group).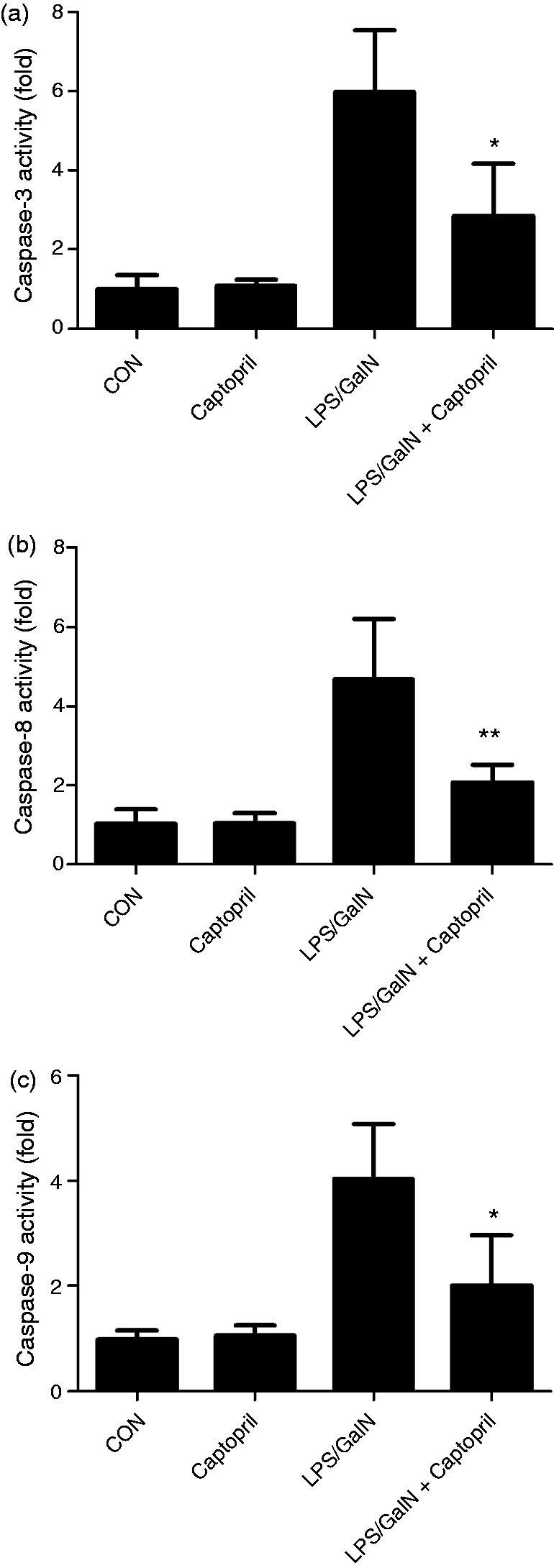
Captopril ameliorated LPS/GalN-induced mortality
The survival curve shown in Figure 8 indicated that mice began to die 6 h after LPS/GalN injection, and mortality reached 85% at 18 h in the LPS/GalN group. Consistently, treatment with captopril significantly reduced the mortality.
Captopril decreased LPS/GalN-induce mortality. Mice were treated with vehicle or captopril in the absence or presence of LPS/GalN challenge. The survival curve of mice with or without captopril treatment and exposed to LPS/GalN (n = 20; **P < 0.01, compared with the LPS/GalN group).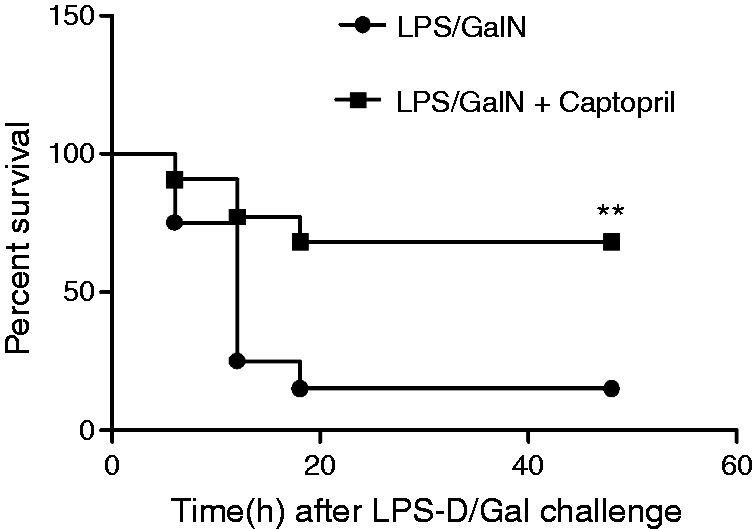
Discussion
Accumulating evidence indicates that the RAS plays extensive and complex physiological function in various organs, 21 while the aberrant activation of the RAS is involved in the development of a series of pathological processes. 22 Therefore, the ACEI and the other reagents targeting the RAS might have comprehensive pharmacological significance in clinical patients.23,24 In inflammation-based disorders, treatment with ACEI helps to control the inflammatory response and alleviate the tissue injury.7,9,11 Previous studies have found that exposure to LPS significantly activated the RAS and increased the concentration of angiotensin II in plasma.25,26 In the present study, we found that administration of captopril, a representative ACEI, attenuated LPS/GalN-induced fulminant liver injury and decreased the mortality of the mice with experimental acute hepatic failure. The present data suggested that ACEI could also provide benefits in lethal liver disorders.
The liver injury induced by LPS/GalN in mice is a classical model widely used in experimental investigation of fulminant hepatitis. 27 The exposure to LPS/GalN stimulates a strong inflammatory response in the liver, and the liver injury largely depends on the excessive induction of the detrimental inflammatory mediators such as TNF-α. 28 The anti-inflammatory actions of ACEI have been well studied both in vitro and in vitro previously. 29 In agreement with these studies, the current experiment found that treatment with captopril suppressed the elevation of TNF-α and IL-6 in LPS/GalN-challenged mice. The suppressed inflammatory response might contribute greatly to the beneficial effects of captopril in LPS/GalN-induced fulminant hepatitis.
Although the pro-inflammatory activities of angiotensin II have been confirmed in vitro and in vivo, the underlying the molecular mechanisms have not been fully elucidated. It has been reported that angiotensin II up-regulated TLR-4 mRNA and protein,30,31 which might be responsible for the pro-inflammatory activity of angiotensin II. 32 In addition, angiotensin II could enhance inflammation via activation of the PKC/NF-κB pathway, promoting transcription factor 3 SUMOylation and inducing ROS generation.33–35
Of these inflammatory mediators, TNF-α plays a pivotal role via propagation of the downstream pro-inflammatory cytokine release and directly activating the extrinsic apoptotic pathway. 36 Several studies have confirmed that TNF-α is the most potent contributor to LPS-induced liver injury because mice without the TNF-α gene or its receptor were resistant to LPS/GalN exposure. 37 In addition to TNF-α, the apoptosis-inducing properties of angiotensin II have been studied for decades. It was reported that angiotensin II could induce apoptosis in cardiomyocytes, alveolar epithelial cells, glomerular epithelial cells and vascular smooth muscle cells.38–41 Therefore, the suppressed activation of capases and reduced TUNEL-positive apoptotic cells in the present study might be a result of the decreased level of pro-apoptotic TNF-α and angiotensin II in the captopril-treated group.
Interestingly, it was reported that TNF-α might induce the de novo synthesis of angiotensin II and block the synthesis or function of angiotensin II-suppressed TNF-α-induced apoptosis in lung epithelial cell and renal tubular epithelial cells.42,43 Thus, angiotensin II might be an indispensable factor that mediates the pro-apoptotic activity of TNF-α and other apoptosis inducers.44,45 Based on this evidence, the suppressed hepatocyte apoptosis might also be attributed to the interrupted activation of angiotensin II-dependent pro-apoptotic pathway of TNF-α.
In conclusion, the present study found that treatment with the ACEI captopril significantly alleviated liver injury and improved the survival rate in mice with LPS/GalN-induced fulminant hepatitis. These protective benefits might result from suppressed production of pro-apoptotic factor TNF-α and angiotensin II. Although the underlying mechanisms and the potential signaling pathway remain to be further investigated, these data suggest that the RAS might be involved in the progression of LPS/GalN-induced fulminant hepatitis and ACEI might have potential value in inflammation-based lethal hepatic disorders.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the grants from the Chongqing Municipal Education Commission (No. KJ1400235).