
Abstract
NK cells play a vital role in innate anti-tumor immunity. Crosstalk between NK cells and dendritic cells (DCs) has come to the forefront in protection against tumors in the context of DC vaccines. We previously discovered that NK cell activation mediates the anti-tumor activity elicited by DC vaccines in response to melanoma tumor challenge in a murine lung metastasis model. In this study, we sought to explore the mechanism behind this NK–DC communication, specifically looking at the involvement of IL-15 and type I IFN signaling. Using DCs from IL-15–/– and IL-15Rα–/– mice, we found that the anti-tumor effect of the vaccine remained comparable with DCs from wild type mice. Moreover, DCs derived from IFN-α/βR–/– mice also maintained their anti-tumor effect. Interestingly, endogenous DCs were found to accumulate in the draining lymph nodes post-immunization and their depletion abolished the anti-tumor effect of the vaccine. Our findings suggest the important role that type I IFN signaling and endogenous DCs play in DC vaccine-mediated anti-tumor protection. Our data suggest that type I IFNs from vaccine DCs activate host DCs to provide NK cell-mediated anti-tumor immunity.
Introduction
NK cells are present in lymphoid and non-lymphoid peripheral tissues, and are involved in innate defense against tumors.1,2 Strategies are emerging to apply NK cells as therapeutic agents against a broad range of malignancies.3,4 NK cells and dendritic cells (DCs) are becoming increasingly recognized as key players in anti-tumor and antiviral immune processes. Regarding anti-tumor activity, NK cells have numerous effector functions, which include granule exocytosis and death receptor engagement, leading to apoptosis, as well as IFN-γ production, which directly affects tumor growth. 5 These effector functions can be activated by type I IFNs, which signal through a heterodimeric receptor consisting of an IFN-α/β receptor 1 (IFN-α/βR1) and an IFN-α/β receptor 2 (IFN-α/βR2). 6
The importance of communication between DCs and NK cells in innate immunology was discovered by Fernandez et al., who found that NK cell effector functions were activated by direct cell-to-cell contact with DCs in a mouse tumor model. 7 Since then, the role of DC vaccines and subsequent NK cell activation has been explored in various studies.8–13 We previously demonstrated that DCs and NK cells communicate following an un-pulsed DC immunization, resulting in tumor protection in an NK cell and IFN-γ-dependent manner. 11
The significance of IL-15 trans-presentation and type I IFN in NK–DC crosstalk has been recognized. IL-15 is a constitutively present, pleiotropic innate cytokine that plays an important role in NK cell activation, proliferation and survival. 14 IL-15 signals through presentation to its trimeric receptor, sharing the IL-2 receptor β and γ subunits, while possessing a unique α subunit. It was demonstrated that IL-15Rα binding to IL-15 was an important factor in maintaining IL-15 stability and bioactivity. 15 Effective intracellular trafficking, subsequent presentation at the cell surface and the secretion of IL-15 by DCs was also shown to be influenced by the presence of IL-15Rα. 16 Moreover, it was discovered that DC trans-presentation of IL-15 on IL-15Rα was required for NK cell activation via direct cell-to-cell contact.16,17 Thus, IL-15 trans-presentation may be involved in DC vaccine-mediated NK activation and tumor protection. Furthermore, Lucas et al. describe a DC-produced type I IFN autocrine signaling pathway, which results first in DC activation and subsequent cell-to-cell communication with and priming of NK cells. 17 Type I IFN was found to up-regulate IL-15/IL-15Rα, which was necessary for NK priming. 17 However, Martinez et al. demonstrate that NK cell effector functions are mediated through direct stimulation of NK cell IFN-α/βR by DC-produced type I IFN. 18
A more recent study has further described the role of type I IFN and IL-15 in NK–DC interactions and tumor protection. 9 It was found that vaccination with virally infected DCs, which responded to infection by type I IFN secretion, was able to provide protection against pre-established tumors in mice. This protection was shown to be dependent on type I IFN secretion and subsequent NK cell activation. However, an in vitro model showed that DC-produced type I IFN was insufficient for NK activation, indicating that cell-to-cell contact between infected DCs and NK cells is required. The study further demonstrated that IL-15 produced by DCs was essential for their direct interaction with and activation of NK cells, leading to tumor protection. Thus, type I IFN signaling and IL-15 trans-presentation in the context of NK:DC interactions could be involved in DC vaccine-mediated NK stimulation and tumor protection.
The present study aims to further investigate the vaccine-mediated activation of NK cells in the context of tumor defense. Employing a mouse experimental lung metastatic tumor model, we explore the role of DC-produced IL-15/IL-15Rα and DC responsiveness to IFN-α/β in NK cell-mediated tumor clearance. Our data indicate that vaccine DC IL-15/IL-15Rα and IFN-α/β signaling are not necessary in NK cell-mediated protection against tumor challenge. However, host DCs accumulate in draining lymph nodes (LNs) following DC vaccine and are essential in providing NK cell mediated anti-tumor immunity initiated by DC vaccine.
Materials and methods
Animals
Six- to 8-wk-old C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA, USA). IL-15–/– and 129Sv2 mice were purchased from Taconic (Hudson, NY, USA). Breeding pairs of IL-15Rα–/–mice (C57BL/6 background) and IFN-α/βR–/– mice (129Sv2 background) were kindly provided by Dr Ma (University of California, San Francisco, CA, USA) and Dr Zinkernagel (University of Bern, Bern, Switzerland), respectively. CD11c.DTR mice were bred in the McMaster Central Animal Facility. IFN-α/βR–/– and IL-15–/– mice were bred in level II housing in the Central Animal Facility at McMaster University. All mice used for experiment were female and 6–10 wk old. All procedures carried out on mice were approved by the McMaster Animal Research Ethics Board and complied with the guidelines set forth by the Canadian Council on Animal Care.
Flow cytometry reagents
The following Abs and fluorescent reagents were purchased from BD Pharmingen (Mississauga, ON, Canada): anti-CD3 (145-2C11), anti-IFN-γ (XMG1.2), anti-NK1.1 (PK136) and anti-CD69 (H1.2F3). Cells were incubated with Fc-block (BD Pharmingen) for 15 min prior to Ab staining. Most staining conditions involved five fluorochromes (FITC, PE, PE-Cy5, PE-Cy7 and APC), and data were acquired using a FACSCanto and analyzed using FlowJo (Tree Star, Ashland, OR, USA).
Cell culture
Murine melanoma B16F10 cell lines were cultured in RPMI medium supplemented with 10% FBS, 2 mM
DC generation from bone marrow cultures
Bone marrow (BM) cells were cultured in the presence of GM-CSF for 7 d as previously described. 19 Cells were fed with fresh medium and cytokine on d 3 and 6 of culture. DCs were matured by 16 h stimulation with 2 µg/ml of CpG.
DC immunization and tumor challenge
Mice were immunized with 1 × 106 DCs or PBS via footpad injection as previously described. 11 All recipient mice were of C57BL/6 background, with the exception of 129Sv2 background mice receiving IFN-α/βR–/– DCs. Tumor challenge in mice was done according to the same model as previously described, with minor modifications. 11 B16F10 melanoma cells (1 × 106) (ATCC) were injected i.v. 7 d after immunization and mice were sacrificed 14 d thereafter.
NK cell depletion
In vivo depletion of NK cells was performed using the mAb PK136 against NK1.1. Purified Abs (100 µg) or isotype control Abs were injected intraperitoneally (i.p.) 2 d before tumor challenge and continued twice weekly until mice were sacrificed for enumeration of lung metastases.
Tumor nodule quantification
Lungs were surgically removed and fixed in 2% paraformaldehyde for 24 h and kept in 70% ethanol before enumeration. The lung metastatic nodules were counted under a dissecting microscope on d 14 after tumor challenge. Tumor nodule digital images were obtained for all lungs under light microscope and digital camera.
Endogenous DC characterization
DCs were cultured as described above and labeled with carboxyfluorescein succinimidly ester (CFSE) to distinguish them from endogenous DCs. C57BL/6 mice were injected with the labeled DCs or PBS via the footpad, as previously described. Popliteal LNs were harvested 48 h post-injection. Single cell suspensions were prepared, counted and stained for CD3, CD11c, CD19, MHCII, CD80 and CD86. Live and single cells were gated on and flow cytometric analysis of CFSE– CD3– CD19– CD11c+cells was done.
CD11c depletion
To deplete CD11c DCs, we used either CD11c.DTR mice or generated chimera by transferring BM cells from CD11c.DTR mice (as donor) into recipient B6 mice.
DTR mouse model
CD11chi DCs were ablated from CD11c.DTR mice by a single injection of 4 ng DT/g body mass via the intraperitoneal route. DC immunization was done 48 h post-DT injection.
BM chimera model
BM cells were isolated from femurs and tibias of CD11c.DTR mice. C57BL/6 mice were irradiated (2 × 550 Rads; 4 h interval). BM cells (9 × 106) were transferred per irradiated mouse via i.v. tail injection. Eight wk post-irradiation, DT was administered. Injection of DT occurred every other day for a total of three injections (4 ng DT/g), beginning 1 d prior to immunization with DCs.
Statistical analysis
Means were compared using one- and two-way ANOVAs, employing Tukey’s and Bonferroni post-hoc tests, respectively. For all tests, P < 0.05 was used to indicate statistically significant differences. All statistical analyses were carried out using GraphPad Prism 5 software (GraphPad, La Jolla, CA, USA).
Results
Functional NK cells are required for anti-tumor immunity provided by DC vaccines
We have shown that DC vaccines are capable of activating host NK cells to exhibit cytolytic activity and IFN-γ production, providing protection against metastatic melanoma.
11
In order to further prove that the DC vaccine protection is NK-mediated, we examined tumor immunity in mice depleted of or genetically lacking functional NK cells. We first performed an in vivo depletion analysis using specific anti-NK1.1 Abs post-DC immunization, but 2 d prior to i.v. challenge with B16F10 melanoma cells. DC-treated animals were largely protected against tumor development, whereas massive tumor nodules could be visualized in the lungs of control mice injected with PBS (Figure 1A). The DC vaccine-induced tumor protection was completely abolished in mice treated with anti-NK1.1 Abs prior to the challenge (Figure 1A). Furthermore, DCs from C57BL/6 or 129Sv2 mice were inoculated into IL-15–/– or IFN-α/βR–/– mice, respectively, which lack functional NK cells. The mice were challenged with B16F10 cells 7 d post-immunization and tumor nodules were enumerated in lungs 2 wk later. DC-treated animals were largely protected, whereas tumor outgrowth was observed to occur in the lungs of IL-15–/– or IFN-α/βR–/– (Figure 1B) mice at levels comparable to PBS-treated animals. These findings support the notion that NK cells play a crucial role in tumor immunity induced by DC vaccines.
Loss of NK cell-mediated anti-tumor immunity provided by DC vaccine in IFN-α/βR–/– and IL-15–/– mice. (a) DC were cultured from BM of C57BL/6 mice and matured overnight with CpG. C57BL/6 or IL-15–/– mice were injected via the footpad with the matured DCs and 7 d later challenged i.v. with B16F10 melanoma cells. NK depletion in C57BL/6 mice using mAb PK136 started 1 d before tumor challenge and then twice per wk for 2 wk. Mice were sacrificed 14 d post-challenge and tumor nodules were counted at the lung surface under a dissecting microscope. Bars represent means ± SEM for 2–4 experiments, with five mice per group. *Significantly different than PBS group (P < 0.001) or NK-depleted mice. (b) DC were cultured from BM of C57BL/6 or 129SV mice and matured overnight with CpG. C57BL/6, IFN-α/βR–/– and IL-15–/– mice were injected with the matured DCs via footpad and 1 wk later challenged i.v. with B16F10 melanoma cells. Mice were sacrificed 14 d later and tumor nodules were counted at the lung surface under a dissecting microscope. Bars represent means ± SEM for 2–4 experiments, with five mice per group. *Significantly different from PBS groups (P < 001). Note: value of 400 assigned when tumor nodules uncountable.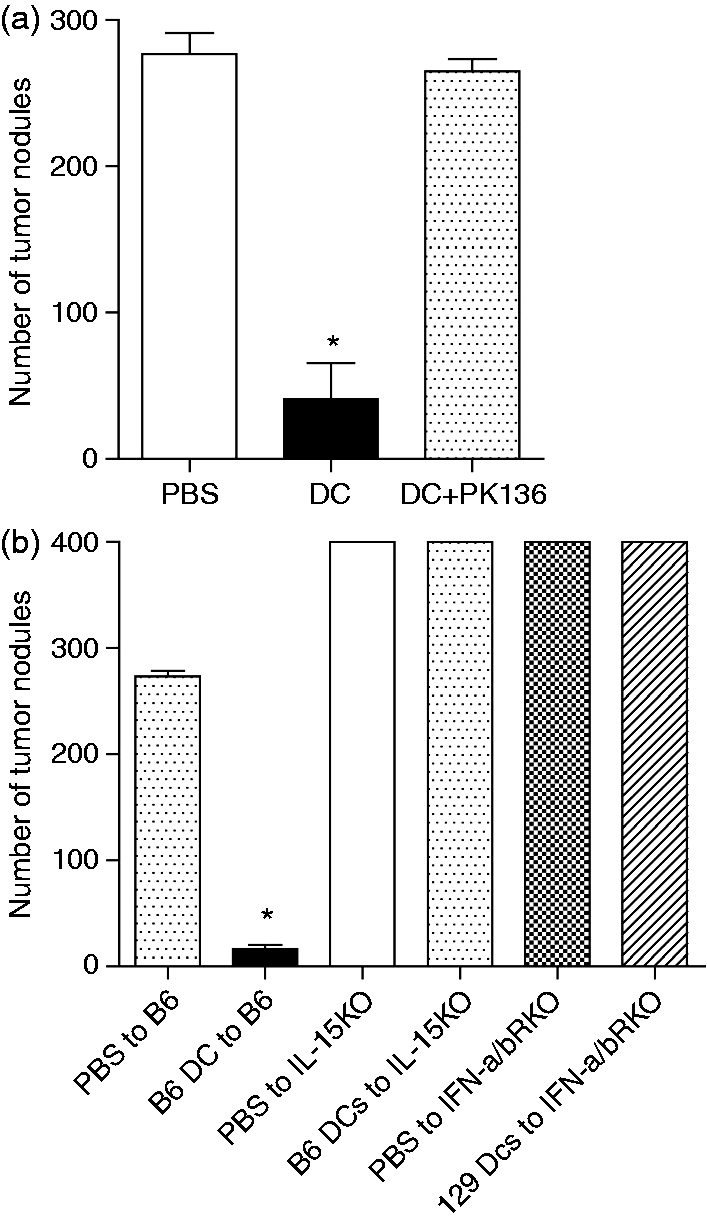
Neither IL-15/IL-15R complex nor IFN-α/βR signaling is required for DC vaccine NK-mediated tumor protection
It has been suggested that trans-presentation of IL-15 by DCs is an important factor in the proliferation and activation of NK cell effector functions. Moreover, the IFN-α/β autocrine signaling pathway has been shown to activate DCs, which subsequently activate NK cells. The NK cell-mediated anti-tumor immunity following DC vaccination demonstrated in the current study prompted us to investigate whether DC vaccines activate NK cells via the IL-15/IL-15R complex and/or IFN-α/βR signaling. C57BL/6 mice received footpad injections of PBS or unpulsed DCs cultured from the BM of C57BL/6, IL-15–/–, and IL-15Rα–/– mice, 7d prior to i.v. challenge with B16F10 melanoma cells. All animals treated with the DCs, which differentiated from genetically different BM, were largely protected against the tumor challenge (Figure 2A, B), whereas tumor outgrowth was observed to occur in the lungs of PBS-treated animals. Furthermore, tumor protection was demonstrated in DC-treated animals where DCs originated from either IFN-α/βR–/– or littermate background 129Sv2 mice (Figure 2C). These findings suggest that NK cell-mediated anti-tumor immunity is independent of DC vaccine-IL-15/IL-15 R complex and the protection does not require vaccine IFN-α/βR signaling pathway.
Neither IL-15/IL-15R complex nor IFN-α/βR signaling is required for DC vaccine NK-mediated tumor protection. (a) DCs were cultured from BM of C57BL/6, IL-15–/– and IL-15Rα–/– mice, and matured overnight with CpG. C57BL/6 mice were injected with the matured DCs via footpad and 7 d later challenged i.v. with B16F10 melanoma cells. Mice were sacrificed 14 d later and tumor nodules were counted at the lung surface under a dissecting microscope. The results are representative of 2–4 experiments, with five mice per group. *Significantly different from PBS groups (P < 0.001). (b) Images of lungs with visible tumor nodules taken post-tumor challenge from C57BL/6 mice injected with PBS, C57BL/6 DCs, IL-15–/– DCs or IL-15Rα–/– DCs. (c) DCs were cultured from the BM of C57BL/6 or IFN-α/βR–/– mice and CpG was used for overnight maturation. C57BL/6 or 129Sv2 mice were inoculated with the DCs and 7 d later challenged i.v. with B16F10 melanoma cells. The mice were sacrificed 14 d after tumor challenge for the enumeration of tumor nodules. Bars represent means ± SEM for 8–10 mice. *Significantly different from PBS groups (P < 0.05). Note: value of 400 assigned when tumor nodules uncountable.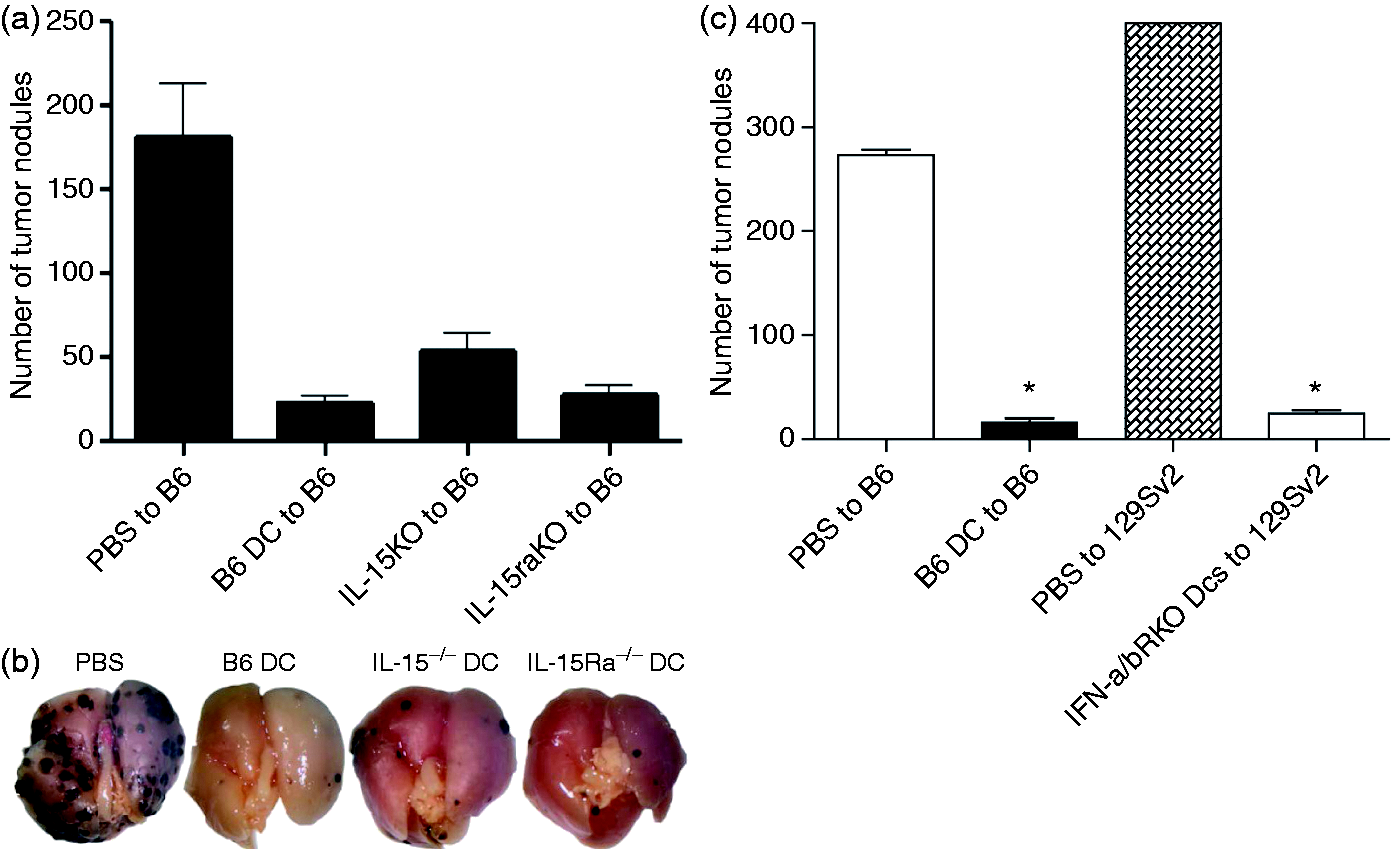
Host DCs accumulate in draining LNs following DC vaccine
We had previously demonstrated that DC vaccines engaged NK cells to trigger their functional maturation and tumor protection.
11
However, surprisingly, our findings ruled out the requirements of the IL-15/IL-15 R complex and IFN-α/βR signaling in the NK-mediated anti-tumor immunity provided by DC vaccine. The findings that NK cell priming is dependent on the recognition of type I IFN signals by DCs, which, in turn, induces IL-15 secretion and IL-15 trans-presentation to resting NK cells, led us to investigate the contribution of host DCs in NK cell stimulation by DC vaccine and tumor protection. We reasoned that when IL-15 signaling is absent in the DC vaccine, owing to either lack of IL-15 secretion or IL-15 receptor expression, the signaling pathways could still be triggered through endogenous DC activation. Indeed, in certain circumstances where DC vaccine lacks IL-15 generation, host DC-derived IL-15 could be taken up by the vaccine to stimulate NK cells. To examine this hypothesis, we performed a series of experiments beginning with characterization of host DCs after administration of DC vaccine. C57BL/6 mice were given a footpad injection of PBS- or CFSE-labeled DCs and 2 d later, the point at which the highest numbers of NK cells accumulate in the draining LNs, the popliteal LNs were harvested and endogenous DCs were phenotypically examined.
11
Flow cytometric analysis of CFSE– CD3– CD19– CD11c+ cells revealed a slight increase in the percentage of CD11c+ cells with a less mature phenotype (Figure 3), expressing lower levels of MHC II and the co-stimulatory molecule, CD86, in the LNs of DC-treated animals in comparison to the controls. Additionally, changes in the cellularity of the draining LNs following DC vaccine led to a huge increase in the numbers of endogenous DCs, increasing from 10,960 ± 2263 to 67,954 ± 17,698 (Figure 3).
Host DCs accumulate in draining LNs following DC vaccine. DCs were cultured from C57BL/6 mice. The cells were treated with CpG, labeled with CFSE and then injected into C57BL/6 mice via the footpad. Popliteal LNs were harvested 48 h post-injection. Single cell suspensions were prepared, counted and stained for CD3, CD11c, CD19, MHCII, CD80 and CD86. (a) Live, single and CFSE– CD3– CD19– CD11c+ endogenous DCs were monitored for expression of MHCII and co-stimulatory molecules. *Significantly different from PBS treated controls (P < 0.05). (b) Graphs represent total number of DCs in two popliteal LNs of mice and percentage of DCs out all cells in the LNs. Bars represent means ± SEM for 4–5 mice. *Significantly different from PBS treated controls (P < 0.05).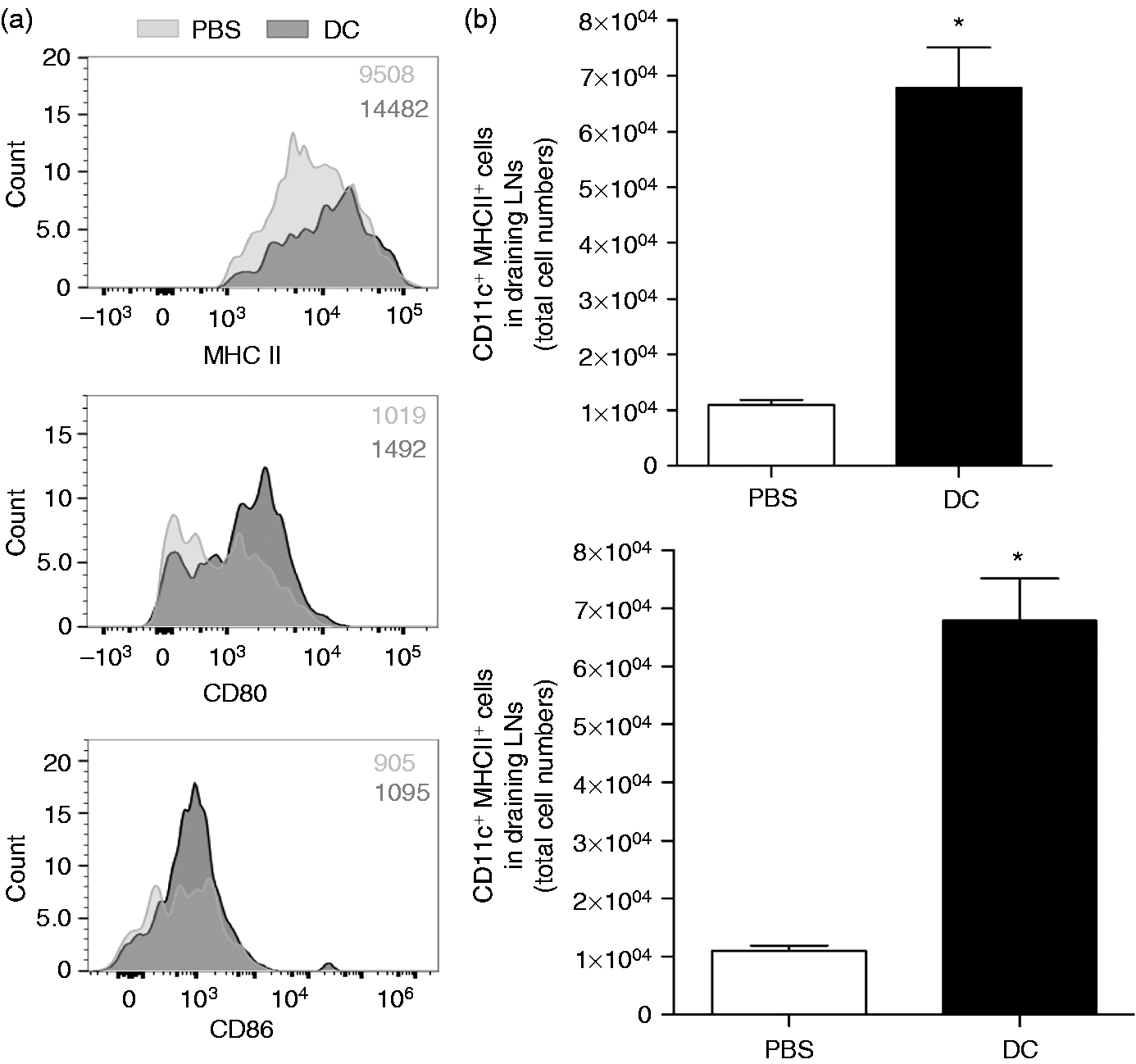
Host DCs are required for NK-mediated protective immunity against tumor challenge
We have previously demonstrated that NK-mediated tumor protection needs at least 3 d post-DC immunization.
11
These observations led us to consider factors other than the DC vaccine in the process of NK engagement and tumor protection. We reasoned that the accumulation of endogenous DCs observed in this study (Figure 3), in parallel with recruitment of NK cells to the draining LNs, could further engage NK cells and provide extra help to these cells to fight the tumor challenge. Therefore, we wanted to determine the effect of host DCs in tumor protection following DC vaccine administration. CD11c.DTR mice were employed and endogenous DCs were depleted prior to the vaccination. CD11c.DTR mice are transgenic mice that express the diphtheria toxin receptor (DTR) under control of the CD11c promoter.
20
DTR mice were administered diphtheria toxin (DT) 48 h prior to DC immunization. The results reveal no protection against tumor challenge after DC immunization in host DC-depleted CD11c.DTR mice (Figure 4A). This illustrates that endogenous DCs are a critical component of the anti-tumor response seen after DC immunization and a lack of host DCs renders the vaccine unable to generate protection against the tumor.
DC vaccine protective immunity against tumor challenge depends on endogenous DCs. (a) Host DCs were depleted from CD11c.DTR mice by one intraperitoneal administration of DT (4 ng/g body mass). DCs were cultured from BM of C57BL/6, IL-15–/– and IL-15Ra–/– mice and matured overnight with CpG. Cultured DCs were injected into the footpad of CD11c.DTR mice 48 h post-depletion, followed by an i.v. B16F10 melanoma challenge 7 d post-immunization. Mice were sacrificed after 14 d and tumor nodules were counted at the lung surface under a dissecting microscope. Bars represent means ± SEM for 3–5 mice. *Significantly different from PBS-treated controls (P < 0.05). In x-axis, (–) indicates administration of PBS. (b) Bone marrow cells from CD11c.DTR mice were used to reconstitute irradiated C57BL/6 mice. DT was administered every other day time, starting 1 d prior to DC immunization. DCs were cultured from BM of C57BL/6, IL-15–/– and IL-15Ra–/– mice and matured overnight with CpG. Chimeric mice were immunized the cultured DCs and challenged with B16F10 melanoma cells 7 d post-immunization. Bars represent means ± SEM for 5–7 mice. *Significantly different from PBS-treated controls (P < 0.05). In x-axis, (–) indicates administration of PBS. Note: value of 400 assigned when tumor nodules uncountable.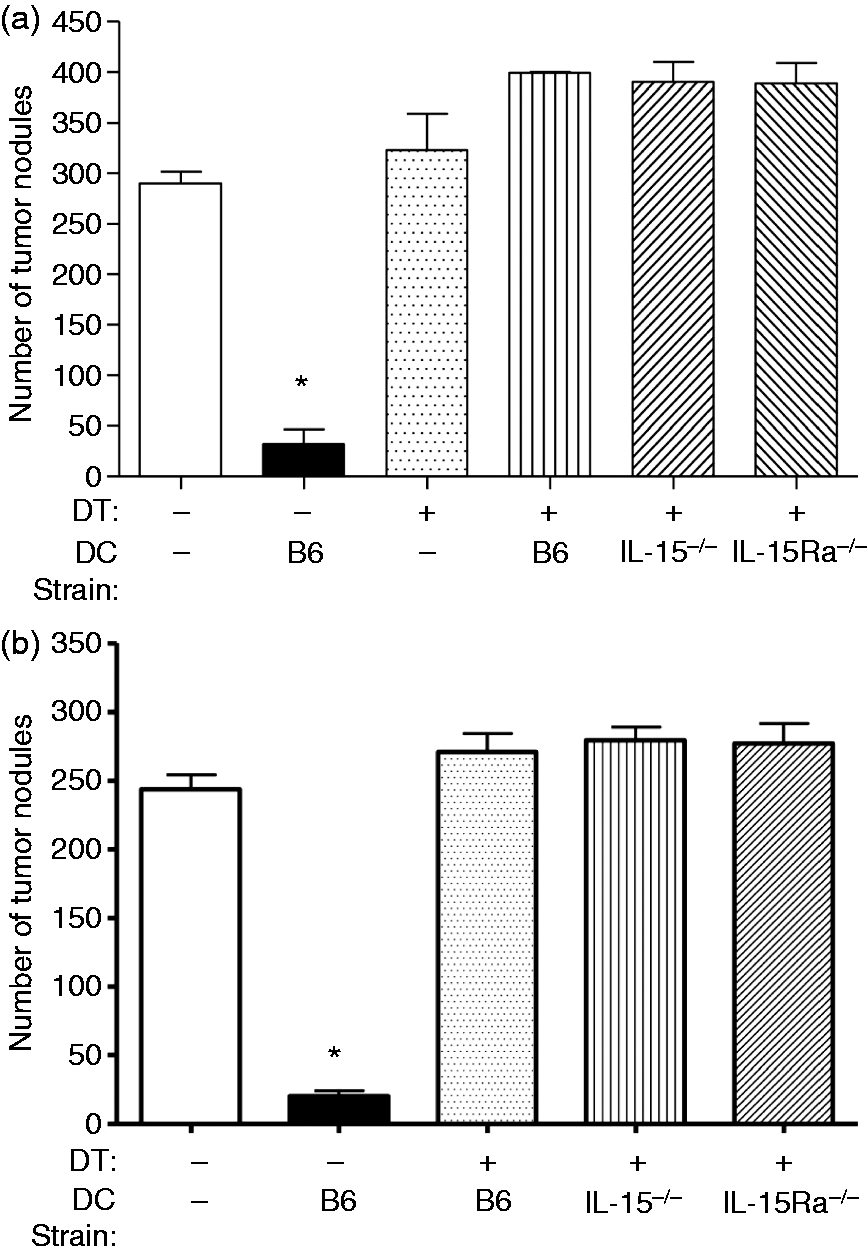
In addition, we generated mixed BM chimeric mice in which endogenous DCs could be selectively depleted without any side effects. BM cells from CD11c.DTR mice were used to reconstitute irradiated wild type recipient mice and DCs were depleted. As seen in the CD11c.DTR mice, tumor growth was observed in the DC-depleted BM chimeric mice after administration of DC vaccine and protection against tumor challenge was absent (Figure 4B). This further illustrates the role of host DCs in immune protection against tumor challenge. The observed anti-tumor immunity provided by DC vaccines is dependent on endogenous DCs and depletion of these cells ceases the protective response.
Discussion
DC vaccines have been explored as a potential cancer immunotherapy and used in clinical trials to treat patients. 21 One such vaccine has already been US Food and Drug Administration-approved for use in certain prostate cancers. 22 The role of CD8+ T cells is well recognized in the field of cancer immunotherapy and their activation tends to be the aim of such therapies. However, NK cells play a significant role in anti-tumor activity and could stand as a target for cancer treatment. 23 NK–DC crosstalk is an important part of this role, and this interaction is widely recognized. However, uncertainties remain about the exact communication that occurs between the two cell types, and we aimed to explore this interaction in the context of DC vaccines to gain further understanding of the resultant anti-tumor immunity.
We previously demonstrated the role of NK cells in un-pulsed DC vaccine-mediated tumor protection in a murine lung metastasis model. Using mice lacking NK cells or lacking functional NK cells (IL-15–/–, NK-depleted and IFN-α/βR–/– mice), we verified that DC immunization is dependent on the activation of NK cells, as the protective effect of the vaccine is absent in these mice. After observing the NK-dependent tumor protection brought on by the DC vaccine, we sought to explore the mechanism behind the NK cell activation and subsequent protection against tumor challenge.
Study findings that aim to explain how NK cells and DCs interact are often contradictory and there is yet to be an agreed-upon mechanism. Among such findings are studies that implicate the role of DC-produced IL-15 and type I IFN, as well as IL-12, IL-2 and IL-18, in NK cell activation.24–29 Some argue that cell-to-cell contact is required and IL-12, IL-15 and IL-18 are not essential for NK cell activation, while others have found that DC-produced soluble factors are solely responsible for the observed activation.7,14,29–31 In the present study, we chose to investigate DC IL-15, IL-15Rα and IFN-α/β signaling, which have been previously explored in other studies.14,16–18 Mortier et al. and Lucas et al. showed that DC IL-15 trans-presentation on IL-15Rα is critical in the activation of NK cells.16,17 IL-15Rα-deficient DCs were unable to prime NK cells in another study. 32 To explore the role of IL-15 signaling, we used BM-derived DCs from IL-15–/– and IL-15Rα–/– mice. Immunization with these DCs revealed no changes to the protective response observed compared with immunization with wild type DCs. Mice were still protected against the melanoma challenge, establishing that neither production nor response to IL-15 is required by the exogenous DCs. In the case of IL-15–/– DCs, it is possible that host-produced IL-15 is taken up and used by the vaccine DCs in order to activate NK cells. However, when IL-15Rα is absent, IL-15 trans-presentation by the exogenous DCs is not possible but a protective effect was still observed. This may suggest a reliance on mechanisms other than IL-15 trans-presentation by the vaccine in order to activate NK cells.
After exploring the role of IL-15, we examined vaccine DC IFN-α/β signaling. A previous study suggested that type I IFN autocrine signaling, leading to DC activation, subsequently leads to NK cell activation, while another demonstrated that type I IFN produced by the DC directly activates NK cell function.17,18 Furthermore, it was found that DCs stimulated with IFN-α were able to activate NK cells, which may have been dependent on IFN-α-dependent up-regulation of the NKG2D ligands MHC class I-related chain A and B. 33 Similarly, Andoniou et al. found NK cell cytotoxicity to be dependent on IFN-α release from DCs. 34 Immunizing wild type mice with DCs from an IFN-α/βR–/– model still led to protection against the tumor challenge at the level of mice immunized with C57BL/6 DCs. This established that the absence of the IFN-α/βR on vaccine DCs led to no changes in its effect. Although type I IFN signaling is required in the host for protection to be seen, these results suggest that type I IFN signaling is not required for the exogenous DCs to exert their effect. Thus, vaccine DCs are not required to respond to type I IFN. However, it may be possible that the vaccine acts by releasing type I IFN, rather than responding to it, rendering the presence of the receptor unnecessary.
After eliminating the role of IL-15 trans-presentation and type I IFN signaling in the mechanism of NK cell activation by the vaccine, we decided to investigate the involvement of host mechanisms that could lead to the observed tumor protection. As the vaccine could still provide protection in the absence of the ability to trans-present IL-15 and respond to type I IFN, we reasoned that there could be a reliance on endogenous DCs. This led us to examine the popliteal LNs for endogenous DC counts. A significant increase in numbers was seen in the draining LNs post-DC immunization compared with mice treated with PBS. This seems to suggest that the vaccine leads to the proliferation of endogenous DCs and their accumulation in the LNs, which may be a critical step in the anti-tumor immunity the vaccine provides. A dependence on host DCs may explain our findings that IL-15 and type I IFN signaling were unnecessary in the vaccine DCs, as these pathways are still intact in the host. To verify the role these DCs may play, we depleted endogenous DCs using CD11c.DTR and chimeric mice. Absence of host DCs completely abolished the protective effect of the DC vaccine and tumor nodule formation exceeded that of PBS-treated mice. These results indicate that exogenous DCs are insufficient for the activation of NK cells and subsequent tumor protection, implicating the role of host DCs in the vaccine’s protective mechanism.
Our findings indicate that endogenous DCs are essential for the observed tumor protection, suggesting an interaction between vaccine and host DCs that results in the activation of NK cells. We found that the vaccine’s effect is independent of IL-15, IL-15Rα, and IFN-α/β signaling, which seemed to contradict several studies that indicate IL-15 trans-presentation on DC IL-15Rα and DC type I IFN signaling are essential for NK cell activation. Considering this along with our own findings of the significance of endogenous DCs, it may be possible that it is not the DC vaccine but the endogenous DCs that require IL-15 or IFN-α/β signaling. Vaccine DCs may act on endogenous DCs through the production of type I IFN, even in the absence of the IFN-α/βR on the vaccine DCs. Activation of endogenous DCs by IFN-α/β could then result in the activation of NK cells. This may be supported by three findings. DCs from wild type mice, which are able to produce type I IFNs and respond to type I IFNs, cannot induce tumor protection in IFN-α/βR–/– mice, where endogenous DCs would lack type I IFN signaling. The inability of endogenous DCs to respond to IFN-α/β could prevent NK cell activation and thus prevent a protective response against the tumor challenge. Moreover, DCs from IFN-α/βR–/– mice, which are able to produce type I IFNs (Gill et al. 35 and unpublished data) but cannot respond to type I IFNs, are able to provide protection against tumor challenge in wild type mice. Finally, depletion of endogenous DCs abrogated the protective effect of injected wild type DCs. Thus, host type I IFN signaling and host DCs are required for DC vaccine-mediated anti-tumor immunity. Although our data clearly show the importance of type I IFN signaling and endogenous DCs in tumor protection elicited by DC vaccines, it would be important to define how vaccine DCs result in the activation of endogenous DCs.
The results of our study further elucidate the communication between NK cells and DCs and the mechanism by which DC vaccines provide anti-tumor immunity. This will serve as an important basis for further clarification of vaccine and endogenous DC interactions, as well as their interaction with NK cells. Moreover, knowledge of these mechanisms provides a foundation for the production of effective anti-tumor therapies. Understanding the communication that occurs between NK cells and DCs, exogenous and endogenous, may allow us to take advantage of the process and further develop and improve DC-based cancer immunotherapy. As DCs and NK cells are also key players in antiviral immunity, the study findings can be extended to infectious models and antiviral therapy.
Footnotes
Funding
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) and Canadian Breast Cancer Foundation (CBCF) to A.A.A.
Conflict of interest
The authors have no potential conflicts of interest to declare.