
Abstract
The O-polysaccharide (OPS, O-Ag) cap of LPS is a major virulence factor of Yersinia species and also serves as a receptor for the binding of lytic bacteriophage φR1-37. Currently, the OPS-based serotyping scheme for the Yersinia pseudotuberculosis complex includes 21 known O-serotypes that follow three distinct lineages: Y. pseudotuberculosis sensu stricto, Y. similis and the Korean group of strains. Elucidation of the Y. pseudotuberculosis complex OPS structures and characterization of the OPS genetics (altogether 18 O-serotypes studied thus far) allows a better understanding of the relationships among the various O serotypes and will facilitate the analysis of the evolutionary processes giving rise to new serotypes. Here we present the characterization of the OPS structure and gene cluster of Y. similis O:9. Bacteriophage φR1-37, which uses the Y. similis O:9 OPS as a receptor, also infects a number of Y. enterocolitica serotypes, including O:3, O:5,27, O:9 and O:50. The Y. similis O:9 OPS structure resembled none of the receptor structures of the Y. enterocolitica strains, suggesting that φR1-37 can recognize several surface receptors, thus promoting broad host specificity.
Introduction
LPS has been identified as a major virulence factor in the Gram-negative pathogen Yersinia pseudotuberculosis.1,2 It also functions in certain Yersinia strains as a receptor for the binding of lytic bacteriophage φR1-37. LPS is composed of lipid A, which anchors LPS to the outer membrane of the bacterial cell wall, core oligosaccharide (core-OS) and O-specific polysaccharide (OPS or O-Ag). The OPS is the most distal component of the LPS, and typically consists of a chain of O-units comprised of various sugar residues linked together by glycosidic bonds in a specific combination. OPS polymorphism within a species is exploited in serology for the classification of strains, and the current O-serotyping scheme for Y. pseudotuberculosis is comprised of 21 different OPS serotypes. 3 The original Y. pseudotuberculosis species was recently reclassified into two distinct species with one retaining the former name and the other named Y. similis. 4 This distinction was confirmed in a multilocus sequence typing (MLST) study 5 where it was proposed that the two species, together with Y. pestis, should be considered as a Y. pseudotuberculosis complex. The study also indicated that while the Y. similis and Y. pseudotuberculosis strains belonged to different MLST types, they shared the O-serotypes between them.
In all Y. pseudotuberculosis serotypes that have been studied so far, the genes required for OPS synthesis are organized into a gene cluster flanked by the hemH and gsk genes. 6 The OPS gene clusters have a promoter region located immediately downstream of the hemH gene, referred to as the JUMPStart sequence (Just Upstream of Many Polysaccharide Starts). 7 The OPS structure of 15 Y. pseudotuberculosis serotypes contains as a branch a rare, immunodominant 3,6-dideoxyhexose (3,6-DDH) sugar residue. 6 To date, 18 of the 21 OPS gene clusters and the corresponding O-unit structures have been determined for the Y. pseudotuberculosis complex, with only serotypes O:8, O:9 and O:13 remaining to complete the data set.
Bacteriophages often target LPS of Gram-negative bacteria as a receptor, and this is also the case with yersiniophage φR1-37, which can infect various members of the genus Yersinia.8–12 The sensitivity of Y. similis O:9 to bacteriophage φR1-37 served as an additional motivation for the OPS structural studies. Bacteriophage φR1-37 was isolated from sewage owing to its ability to infect OPS-negative strain YeO3-c-R1 of Y. enterocolitica.
12
The φR1-37 receptor in YeO3-c-R1 was localized to the outer core oligosaccharide of LPS, which is composed of Galp, N-acetyl-galactosamine (GalpNAc) and 2-acetamido-2,6-dideoxy-
We present here the branched tetrasaccharide structure of the OPS repeat unit of Y. similis serotype O:9 strain R708 and the organization of the gene cluster that directs the biosynthesis of the OPS.
Materials and methods
Bacterial strain and cultivation
Yersinia similis serotype O:9 strain R708 sensitive to bacteriophage φR1-37 was used in these studies. 8 Fermentor cultivation of bacteria was performed in a BIOSTAT C fermentor at 30℃ with stirring (300–375 rpm), and pH 6.8 was maintained by addition of concentrated ammonium hydroxide as needed. The bacteria were cultivated in Yersinia broth containing tryptone (10 g/l), yeast extract (5 g/l), NaCl (5 g/l), CaCO3 (0.25 g/l), K2HPO4 (7.5 g/l), Glc (10 g/l) and biotin (1 mg/l). Glc was supplemented to the fermentor during the cultivation to a final concentration of 86.3 g/l. After 47.5 h of cultivation the bacteria were killed with 1% phenol, washed twice with 0.9% NaCl and freeze-dried.
LPS isolation, purification and degradation
To isolate LPS, the dry bacterial mass was subjected to hot phenol/water extraction. 13 Collected water and phenol phases were extensively dialyzed in deionized water until free of phenol, and freeze-dried. The water phase containing most of the LPS was further purified by treatment with enzymes and ultracentrifugation. The LPS, dissolved in buffer containing Tris (6.057 g/l), MgCl2·6H2O (0.2 g/l) and with NaN3 (0.02%) to inhibit the bacterial or fungal growth, was digested with RNase (0.02 mg/mg of LPS) and Benzonase (0.5 µl/mg LPS; Sigma-Aldrich; Munich, Germany) for 12 h at 37℃. Then, proteinase K was added (0.01 mg/mg of LPS) and the LPS was further incubated at for 16 h at 37℃ and for 2 h at 60℃. The material was dialyzed against deionized water and freeze-dried. Finally, the purified LPS was sedimented from a 1–3% solution of LPS by ultracentrifugation (105,000 × g, 4℃, 22 h).
The LPS was hydrolyzed in aqueous acetic acid (1%) for ∼ 4 h at 100℃ to cleave the lipid A, then sedimented in an ultracentrifuge (105,000 × g, 4℃, 4 h). The supernatant containing the OPS was fractionated via size exclusion chromatography (SEC) utilizing a column (2.5 × 75 cm) of Sephadex G-50 and 0.05 M pyridinium acetate buffer (pH 4.5) as an eluent. The high molecular mass fraction containing the OPS was chosen for further studies.
The OPS (5 mg) was O-deacetylated with aqueous 12.5% ammonia for 16 h at 37℃ after which the sample was neutralized to pH 7 with acetic acid and desalted on a column (1.5 × 100 cm) of Sephadex G-10 with 0.1 M NH4HCO3 as eluent.
For Smith degradation,14,15 the native LPS (10.5 mg) and OPS (10.2 mg) were oxidized with freshly prepared 0.1 M NaIO4 at 20–22℃ (room temperature) for 48 h in the dark, reduced with NaBH4 (room temperature, 48 h), acidified with acetic acid to pH ∼ 5 and applied to gel permeation chromatography (GPC; Sephadex G-10). The oxidized material was further hydrolyzed with 1% acetic acid (100℃, 2 h), cooled down, neutralized with NaOH and separated in GPC (Sephadex G-10).
General and analytical chemistry methods
For preliminary chemical composition, the LPS samples were subjected to methanolysis (2 M HCl/MeOH, 85℃, 2 h). After acetylation with pyridine and acetic anhydride (85℃, 10 min) the derivatives were detected by gas chromatography–mass spectrometry (GC–MS). The utilized gas chromatograph was a Hewlett Packard HP 5890 (series II) equipped with a fused-silica SPB-5 column (30 m × 0.25 mm × 0.25 µm film thickness; Supelco, Sigma-Aldrich Group; Munich, Germany), flame ionization detector (FID) and a MS 5989 A mass spectrometer with vacuum gauge controller 59827 A. The temperature program was initiated by 150℃ for 3 min, then the temperature was increased by 5℃/min until a temperature of 330℃ was reached.
Sugar components were also identified as alditol acetate derivatives 16 after hydrolysis (2 M trifluoroacetic acid, 120℃, 2 h), reduction (NaBH4, 16 h in the dark) and acetylation (85℃, 10 min) by GC [HP 5890 series II gas chromatograph with FID and a column (30 m × 2.5 mm × 0.25 µm; Agilent Technologies; Santa Clara, CA, USA) of polysilican SPD-5]. Helium was a carrier gas (70 kPa), and the temperature program was the same as that described above (150℃ for 3 min and increased by 5℃/min until 330℃ reached).
The absolute configuration of the sugars was determined as described previously,
17
as was methylation analysis.18,19 To determine the absolute configuration, standards of Gal and 2-amino-2-deoxy-glucose (GlcN) were utilized. To detect the glucosaminuronic acid configuration it was reduced and compared with authentic
LPS reduction experiments were performed with NaBH4 and hydrazine (30 min at 37℃). The NaBH4 reaction was stopped with 2 M HCl, while the hydrazine was neutralized with acetone. Both samples were methanolyzed, acetylated and analyzed by GC–MS.
MS
Electrospray ionization (ESI) Fourier-transformed ion cyclotron resonance MS was performed in negative ion mode utilizing an APEX Qe (Bruker Daltonics; Bremen, Germany) with a 7-Tesla actively shielded magnet and a dual ESI/Matrix–assisted laser desorption/ionization ion source. Analyzed samples were dissolved in a mixture of 2-propanol, water and triethylamine (50:50:0.001; v/v/v) at a concentration around 10 ng/µl and were sprayed at a flow rate of 2 ml/min. Capillary entrance voltage was set to 3.8 kV, and the dry gas temperature was 200℃. The mass spectra were charge-deconvoluted and mass numbers given refer to the mono-isotopic masses of the neutral molecules.
NMR spectroscopy
Structural analysis of OPS was achieved by different NMR experiments, including one-dimensional 1H-, 13C- and 31P-NMR; two-dimensional (2D) homonuclear techniques of correlation spectroscopy (1H,1H–COSY), total correlation spectroscopy (1H,1H-TOCSY), nuclear Overhauser effect spectroscopy (1H,1H-NOESY), rotating frame Overhauser effect spectroscopy (1H,1H-ROESY), and 2D heteronuclear single-quantum correlation-distortionless (1H,13C-HSQC-DEPT), coupled 1H,13C-HSQC, heteronuclear multiple-bond correlation (1H,13C-HMBC) and heteronuclear multiple quantum coherence (1H,31P–HMQC). Experiments performed after H-2H exchange of the sample with 99.9% 2H2O were recorded at 50℃ with a Bruker DRX Avance 600 MHz spectrometer (operating frequencies of 600.31 MHz for 1HNMR, 150.96 MHz for 13C NMR and 243.01 MHz for 31P NMR), equipped with a 5 mm QXI multinuclear-inverse probe head with a z gradient, and applying standard Bruker software. Chemical shifts were reported relative to an external standard of acetone (δH 2.225, δC 31.45) and 85% H3PO4 (δP 0.00).
PAGE and Western blotting
For the LPS profile analysis, LPS was separated by SDS-PAGE using a 5% stacking and 15% separating gel. The separation was run at a constant voltage of 151 V. The LPS of Salmonella enterica sv. Montevideo SH94 22 and Y. enterocolitica O:3 strain Ye75S 23 were used as smooth heteropolymeric and homopolymeric OPS controls, respectively. The LPS of Y. enterocolitica O:3 strains YeO3-c-R1 23 and Ye75R 24 were used as rough LPS controls.
After SDS-PAGE the material was visualized by silver staining 25 or transferred to a polyvinylidene fluoride membrane (pore size 0.45 µm; Merck Millipore; Darmstadt, Germany). The membrane was soaked in methanol prior to use. Transfer of the LPS to the membrane occurred for 18 h at 4℃ at constant voltage of 30 V. Subsequently, the membrane was blocked in 10% skimmed milk in blotting buffer (50 mM Tris/HCl, 0.2 M NaCl, pH 7.4) for about 1 h at room temperature. Abs diluted in blocking buffer were added and incubated for 16 h. The membrane was washed five times (5 × 5 min) with blotting buffer, and alkaline phosphatase-conjugated goat anti-rabbit or goat anti-mouse Ab in blotting buffer was added in dilution of 1:1000. Incubation (2 h) was followed with washing as before. Finally, the substrates 5-bromo-4-chloro-3-indoyl phosphate and p-toluidine p-nitro blue tetrazolium chloride (Bio-Rad; Munich, Germany) were added as indicated in the supplier’s instructions. Staining was performed in the dark for 15 min, and the reaction was stopped by washing the membrane with ddH20.
In this work, the polyvalent rabbit antiserum against Y. enterocolitica O:3 strain YeO3-c-R1 and mAb 2B5 26 specific to the outer core of this strain were used. The dilutions were 1:100 for antiserum and 1:1000 for mAb 2B5. Alkaline phosphatase-conjugated goat anti-mouse or anti-rabbit IgG were purchased from Dako (Glostrup, Denmark). LPS from Y. enterocolitica O:3 strain YeO3-c-R1 was used as a positive control. 23
Nucleotide sequence analysis
The genomic DNA of Y. similis O:9 strain R708 was isolated using the JETFLEX Genomic DNA Purification Kit (Genomed; Löhne, Germany). Isolated DNA (3 µg) was fragmented and ligated to adapters. Paired-end sequencing was performed on Illumina HISeq 2000 sequencer (Illumina, San Diego, CA, USA) with the read length of 70 nucleotides resulting in 44-fold coverage. Two different approaches were used to generate the draft genome sequence. First, the obtained sequencing reads were aligned against the Y. similis strain N916Ysi genome (accession number ERS008562) using the Bowtie2 short read aligner 27 provided in Chipster v2.5 (CSC – IT Center for Science). The SAMtools package 28 was then used to export the consensus contigs of Y. similis R708 formed by the sequence reads that aligned to the N916Ysi genome. In the second approach, the raw sequence reads of R708 were filtered for quality and subjected to de novo assembly using the Velvet program. 29 Contigs obtained from the consensus and the de novo assembly were combined using GAP4 program in the Staden Package. 30 Based on the similarity with known OPS clusters, the contig containing the complete OPS gene cluster of Y. similis R708 was identified. The 20,670 base pair sequence was deposited via the European Bioinformatics Institute to sequence data bases under accession number AJ539157.
Bacteriophage inhibition assay
The phage inhibition assay was performed as described elsewhere. 26 Briefly, lyophilized native and Smith-degraded Y. similis O:9 LPS preparations were diluted into ddH20 at the concentration of 1 mg/ml. Serial dilutions of LPS were treated with bacteriophage φR1-37 as follows. Approximately 200–500 infecting phage particles [plaque-forming units (PFU)] were diluted in buffer and mixed with different concentrations of LPS preparations. LPS of Y. enterocolitica O:3 was used as a positive control. After 30 min incubation at room temperature, the number of PFUs was estimated according to a standard protocol using 0.4% (w/v) soft agar and Y. enterocolitica strain YeO3-c-R1 as the host.31,32
Results
LPS isolation and SDS-PAGE analysis
LPS from fermentor-grown cells of Y. similis serotype O:9 strain R708 (87.9 g) was extracted by the hot phenol/water extraction protocol.
13
A crude LPS yielded 4.1% of the dry bacterial cell mass. Purified LPS (1.7% of dry bacterial cell mass) was obtained by the enzymatic treatments and ultracentrifugation that significantly decreased the nucleic acids and protein contamination. In the silver-stained SDS-PAGE gel, the LPS-banding profile of Y. similis O:9 (Figure 1, lanes 1–3) was very similar to that of the smooth LPS of S. enterica containing a heteropolymeric OPS (Figure 1, lane S1), but distinct from that of the three Y. enterocolitica O:3 strains (Figure 1, lanes S2–S4).
LPS profile of Y. similis O:9 strain R708 in SDS-PAGE analysis and silver staining. Lane S1, S. enterica sv. Montevideo SH94 LPS; lane S2, Y. enterocolitica O:3 strain Ye75S LPS, lane S3, Y. enterocolitica O:3 strain Ye75R LPS, lane S4, Y. enterocolitica O:3 strain YeO3-c-R1 LPS; lanes 1-3, Y. similis O:9 LPS after hot phenol/water extraction (1), enzymatic treatment (2) and purified LPS (3). Preparations applied in amount of 2.5 µg except for lane S2 (7 µg).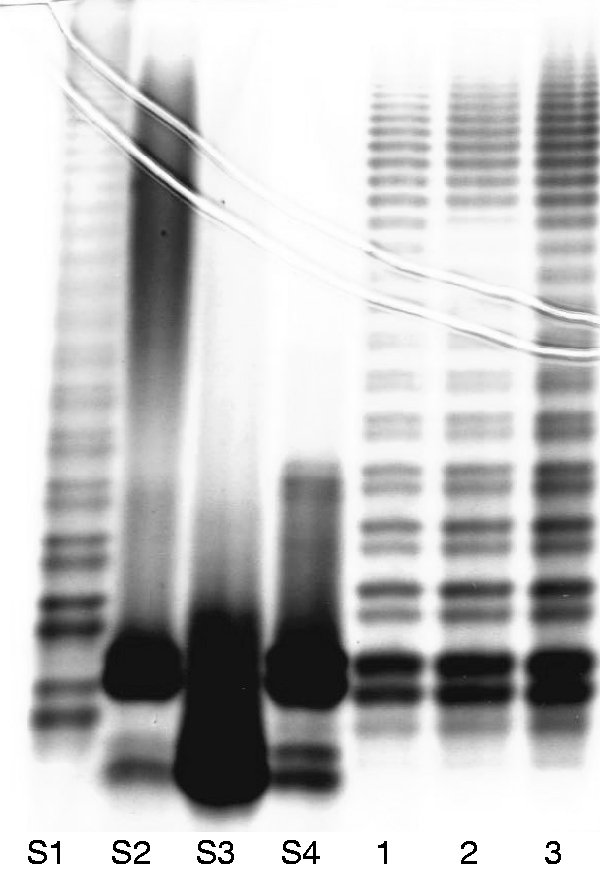
Chemical composition of LPS
As the characterization of the OPS structure was the major goal, purified LPS from Y. similis O:9 was subjected to mild acid hydrolysis and further fractionated in SEC. The first elution fraction with high molecular mass containing OPS and core region (core-OPS) was subjected to further chemical and structural studies (Figure 2).
Gel permeation chromatography (Sephadex G-50) of hydrolyzed Y. similis O:9 LPS (116.6 mg) with aqueous eluent buffer containing 0.4% pyridine and 1% acetic acid. Core-OPS: core with O-polysaccharide; core-OS: core oligosaccharide.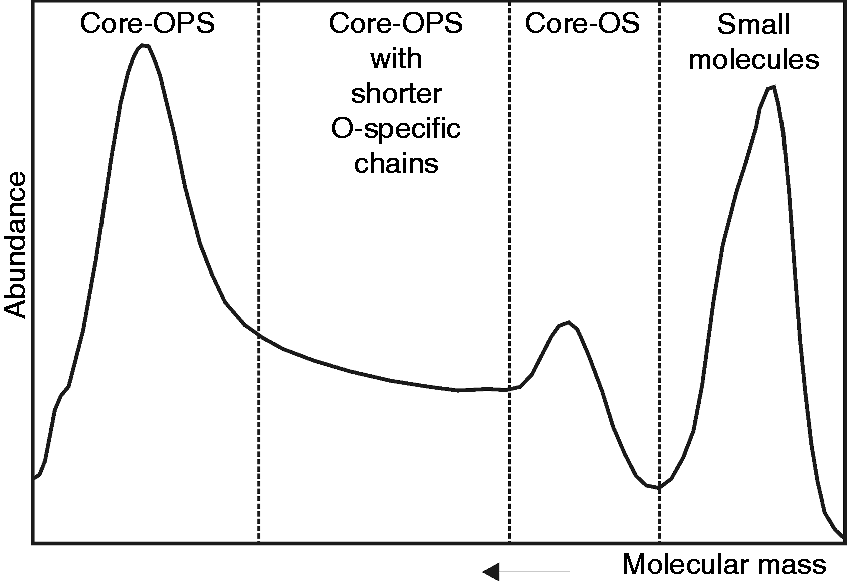
To establish the preliminary chemical composition, the native LPS was analyzed via GC–MS after mild methanolysis and as alditol acetate derivatives. The predominant components were identified as FucN, 4-amino-4-deoxy-arabinose (Ara4N), two different hexoses (Gal, Glc), hexosaminuronic acid identified as glucosaminuronic acid (GlcNA), GlcN, heptose (Hep), 3-deoxy-
The absolute configurations of the sugars considered as OPS components were defined by GC analysis of their acetylated (+)-2-butyl alditols, and identified as
Methylation analysis
For linkage pattern analysis, partially methylated alditol acetate derivatives of OPS were analyzed by GC–MS, which identified 2-N,N-AcMe-1,3,5-tri-O-Ac-2-deoxy-1-deutero-4,6-di-O-Me-glucitol and 2-N,N-AcMe-1,3,4,5-tetra-O-Ac-2,6-dideoxy-1-deutero-galactitol, indicating the presence of 3-substituted GlcpN and 3,4-disubstituted FucpN, 1,5-di-O-Ac-1-deutero-2,3,4,6-tetra-O-Me-galacitol (terminal Galp) and 2-N,N-AcMe-1,3,4,5,6-penta-O-Ac-2-deoxy-1,6,6-tri-deutero-glucitol (3,4-di-substituted GlcpNA, reduced to aminoglucitol).
MS
The ESI Fourier transform (FT) MS analysis of native LPS was unsuccessful as the molecule was too large for measurements. However, the isolated core-OPS fraction yielded convincing results revealing not only the ions corresponding to the core region, but identifying 1–3 residues of the OPS repeating unit as well (Figure 3). In the mass spectrum, the molecular ion at 1120.36 u corresponded to the core region comprised of three heptose residues, two hexoses known to be Gal and Glc, and one Kdo. A second Kdo was not detected, likely owing to its cleavage in acetic acid hydrolysis. Core region heterogeneity was observable as Gal was replaced by a heptose resulting in a mass difference of 30 u. Molecular ions at 1930.66 u and 1978.690 u were consistent with a core oligosaccharide plus a single OPS unit, of which the latter possessed four heptoses and one additional water molecule. Analyzing the ions with decreased masses, the mass differences matched perfectly to an acetyl group (Ac, 42 u), GlcNAcA (217 u), Gal (161 u), FucNAm (187 u) and GlcNAc (203 u). Generally, the ESI FT MS experiment on Y. similis O:9 core-OPS confirmed the compositional studies and demonstrated the presence of acetyl groups, which was further investigated in NMR studies.
Negative ion mode ESI FT MS mass spectrum (deconvoluted) of Y. similis O:9 LPS. The fraction containing the core oligosaccharide with short OPS chain was used as a sample.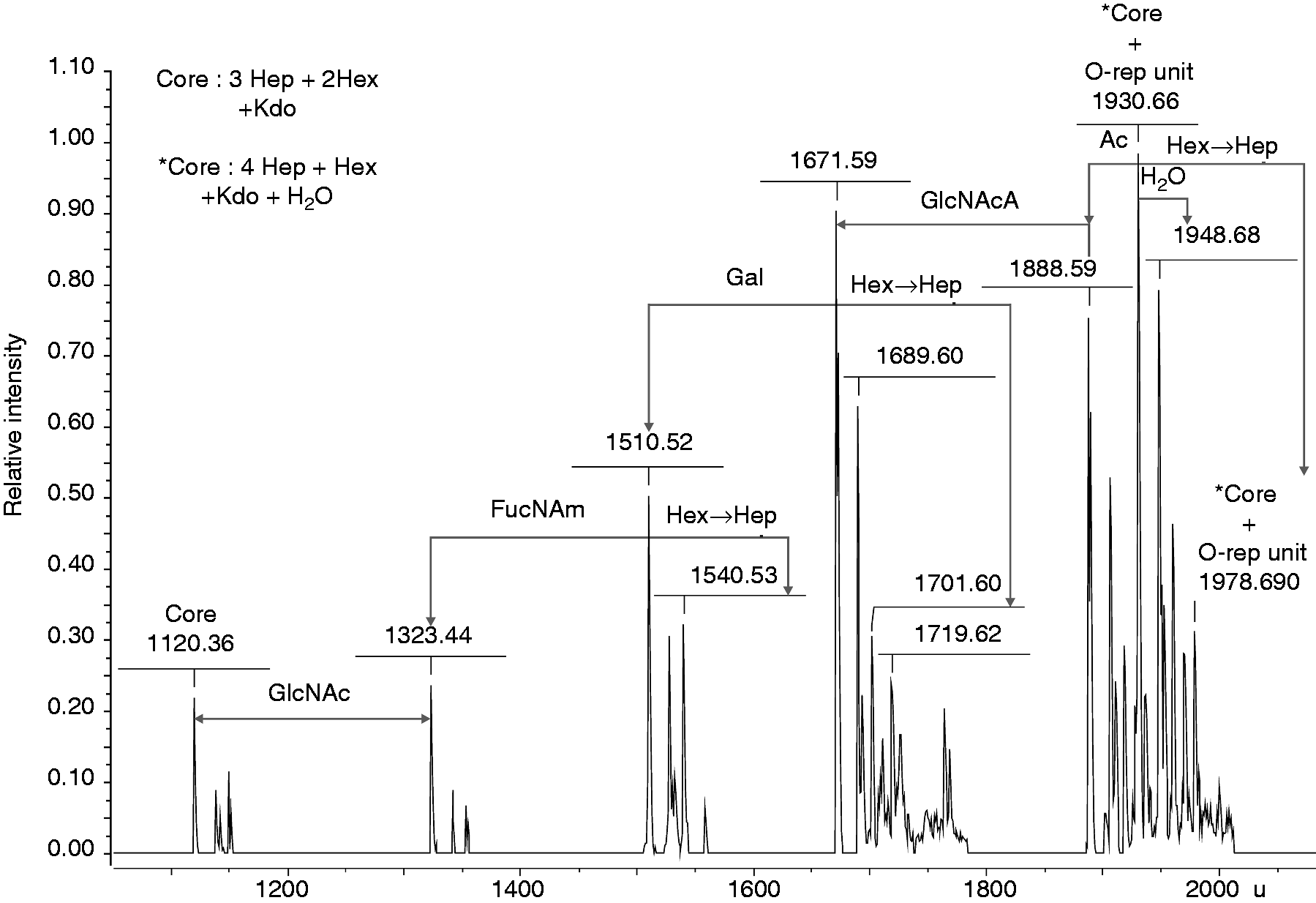
NMR analysis
The structural analysis of the OPS by 13C NMR (Figure 4) showed signals for four anomeric carbons ( 13C NMR spectrum of the OPS isolated from Y. similis O:9. The letters refer to the sugar residues as defined in Table 1 and the arabic numerals refer to the carbons of the respective residues. The spectrum was recorded at 600 Hz and 50℃.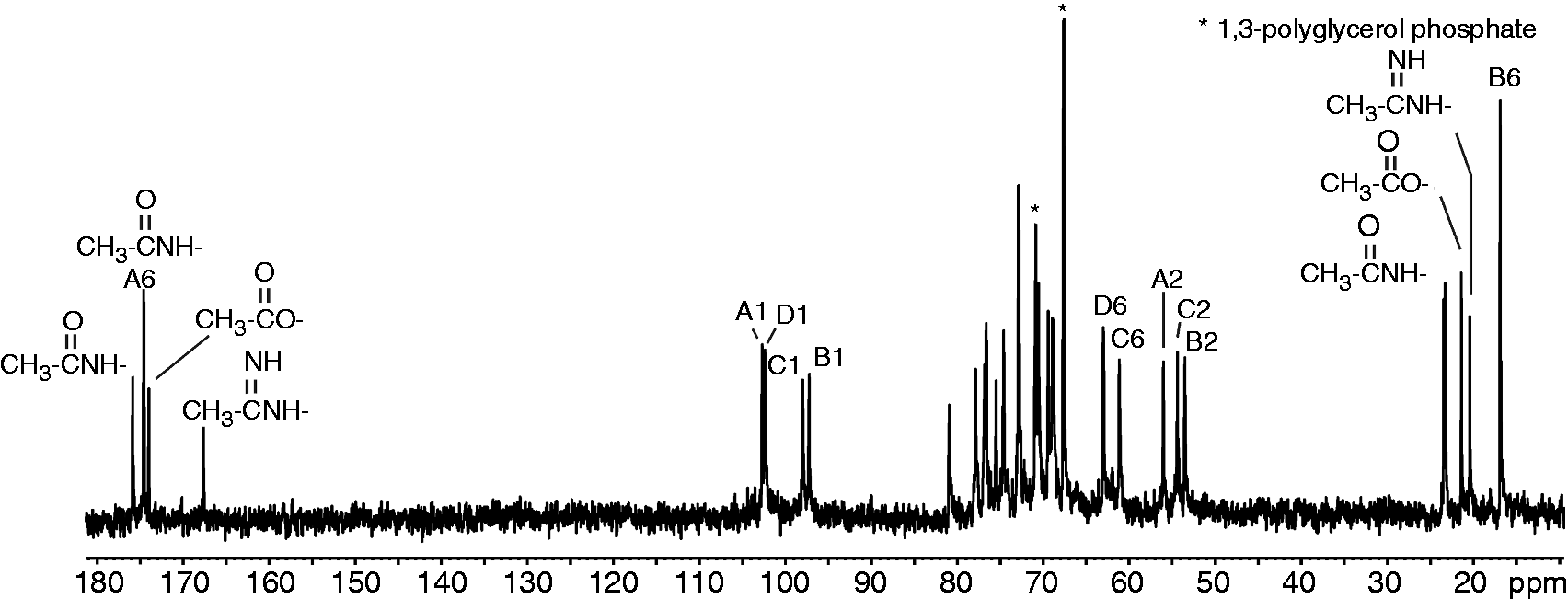
Accordingly, the 1H NMR spectrum (Figure 5A) contained, among other things, five signals at δ 4.81–5.27, which belong to four anomeric protons ( 1H NMR spectra of the OPS isolated from Y. similis O:9. (A) Native OPS. (B) The Smith-degraded OPS. The letters refer to the sugar residues as defined in Table 1 and the arabic numerals refer to the protons of the respective residues. The spectrum was recorded at 600 Hz and 50℃.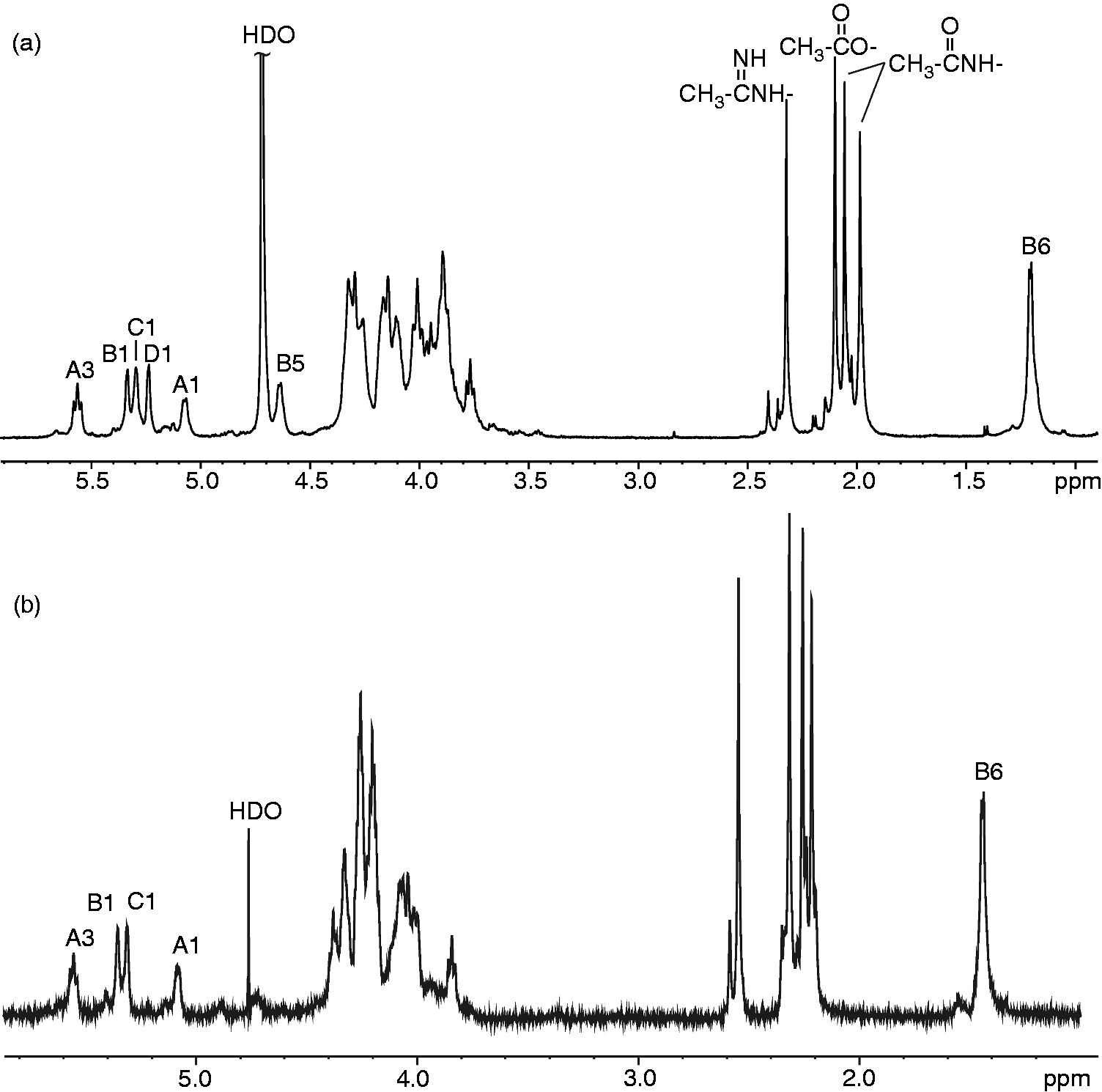
1H and 13C NMR chemical shifts (δ, ppm) of OPS isolated from Y. similis O:9. Spectra were recorded at 50℃ in 2H2O relative to internal acetone (δH 2.225; δC 31.45). Underlined chemical shifts indicate substituted positions.
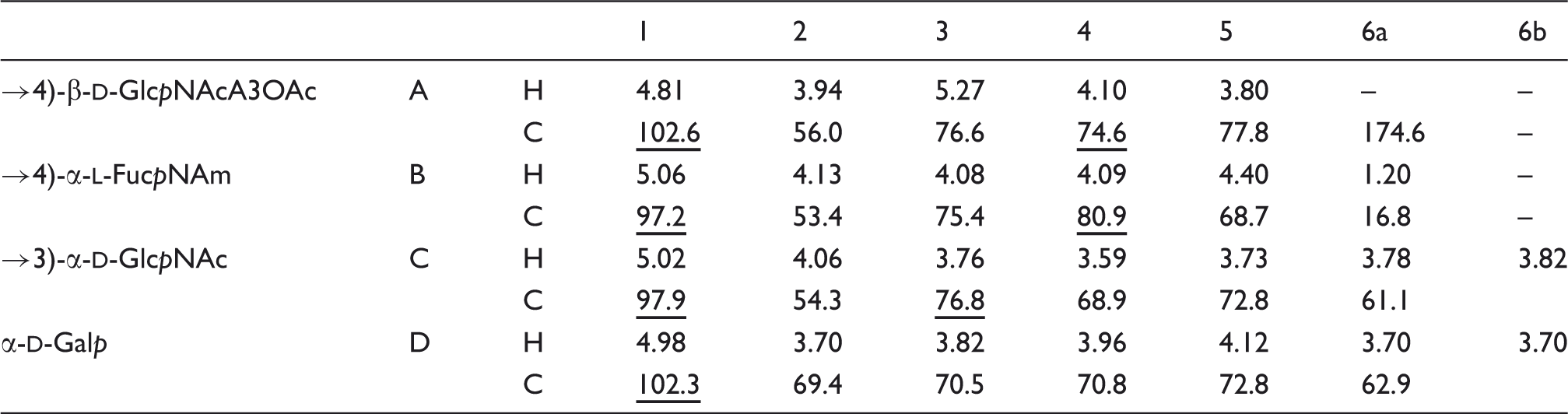
1H and 13C NMR chemical shifts (δ, ppm) of O-deacylated OPS isolated from Y. similis O:9. Spectra were recorded at 50℃ in 2H2O relative to internal acetone (δH 2.225; δC 31.45). Underlined chemical shifts indicate substituted positions.
The NOESY spectrum of the O-deacetylated core-OPS showed the following inter-residue cross-peaks between anomeric protons and protons at the linkage carbons: Structure of the OPS repeating unit in native (a) and O-deacetylated (b) LPS from Y. similis O:9.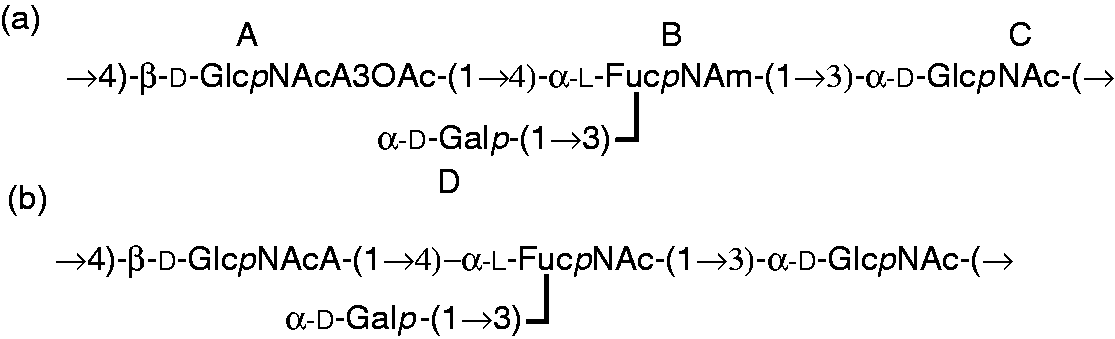
Phage φR1-37 receptor in Y. similis O:9 LPS
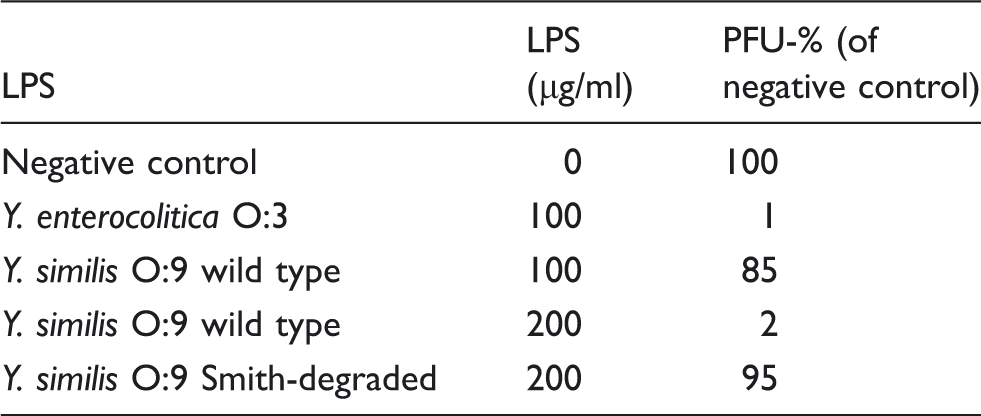
Ability of Y. similis O:9 wild type and Smith-degraded LPS to neutralize phage φR1-37. Y. enterocolitica O:3 LPS was used as positive control.
To further elucidate the possible structural relationship between the Y. enterocolitica O:3 and the Y. similis O:9 LPS, the latter was analyzed in Western blotting with antiserum against YeO3-c-R1 and mAb 2B5 specific for the Y. enterocolitica O:3 outer core. The YeO3-c-R1 antiserum recognized Y. similis O:9 LPS structures, but cross-reactivity was observed only at the low molecular region (Figure 7A), while mAb 2B5 did not react at all with the Y. similis O:9 LPS (Figure 7A).
Western blotting analysis of Y. similis O:9 strain R708 LPS with (a) polyvalent YeO3-c-R1 antiserum (1:100) and (b) mAb 2B5 (1:1000). Lane S4, Y. enterocolitica O:3 strain YeO3-c-R1 LPS (15 µg); lanes 1–3, Y. similis O:9 LPS (30 µg) after hot phenol/water extraction (1), enzymatic treatment (2) and purified LPS (3). LA: Lipid A.
OPS gene cluster sequence analysis
The OPS gene cluster of Y. similis serotype O:9 strain R708 is located between the hemH and gsk genes similar to that of other Yersinia species.6,38,39 Within the O:9 cluster, we identified 20 open reading frames (Orf) of which 16 were predicted to be involved in OPS biosynthesis (Figure 8; Table 4). The presence of a JUMPStart sequence between the hemH and wbuB genes is consistent with observations in different Y. pseudotuberculosis serotypes and the location of the wzz gene immediately upstream of the gsk gene at the 3’-end of the cluster. However, in line with the distinct structure of the repeat unit (Figure 6), the OPS gene cluster shared no similarity with other Y. pseudotuberculosis complex OPS gene clusters.
Organization of the Y. similis O:9 OPS gene cluster and comparison to that of E. coli O145. Figure is drawn to scale. The genes are indicated by arrows, the biosynthetic genes with black, the GTase genes with gray, the OPS assembly protein genes with vertical-striped and other genes with white arrows. The amino acid sequence identity percentages between the gene blocks are indicated between the gene clusters. Note that the Wzx and Wzy sequences of the two organisms share no significant similarity. Y. similis O:9 OPS gene cluster genes and predicted functions.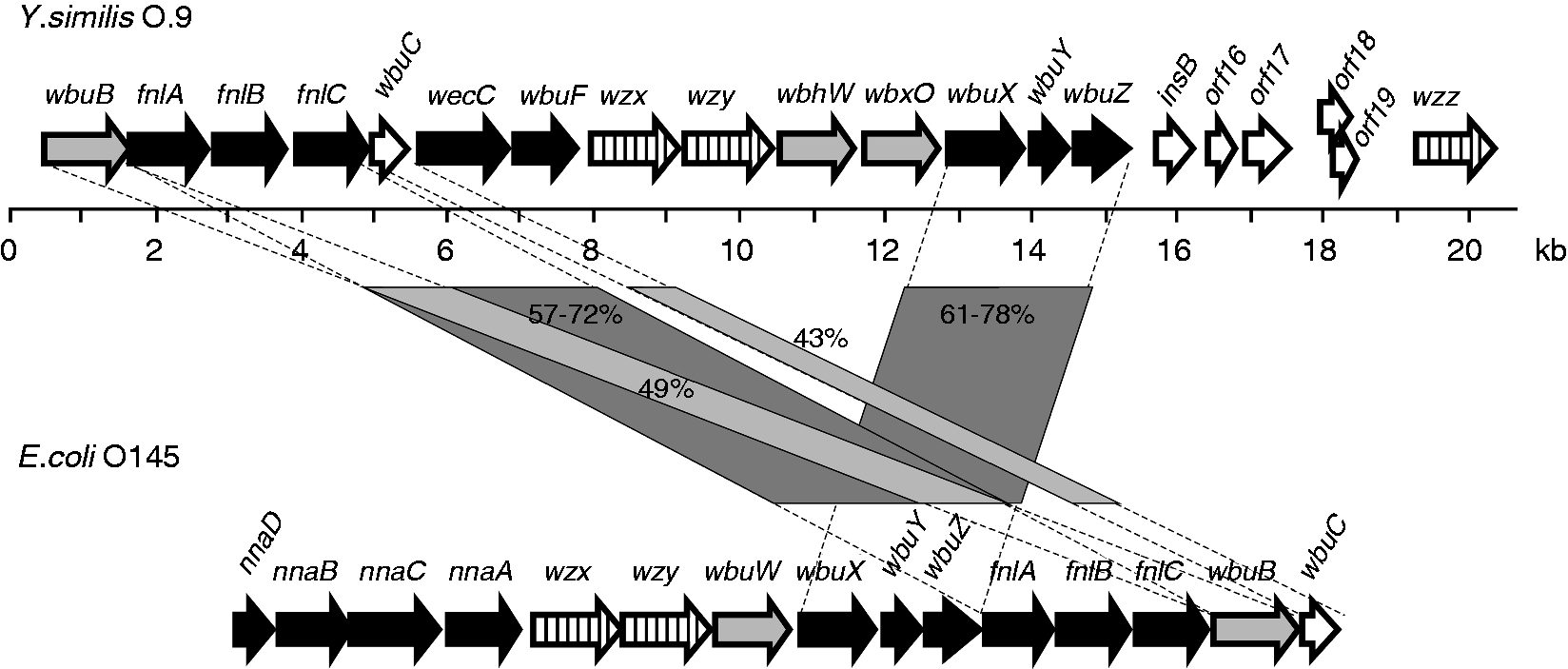
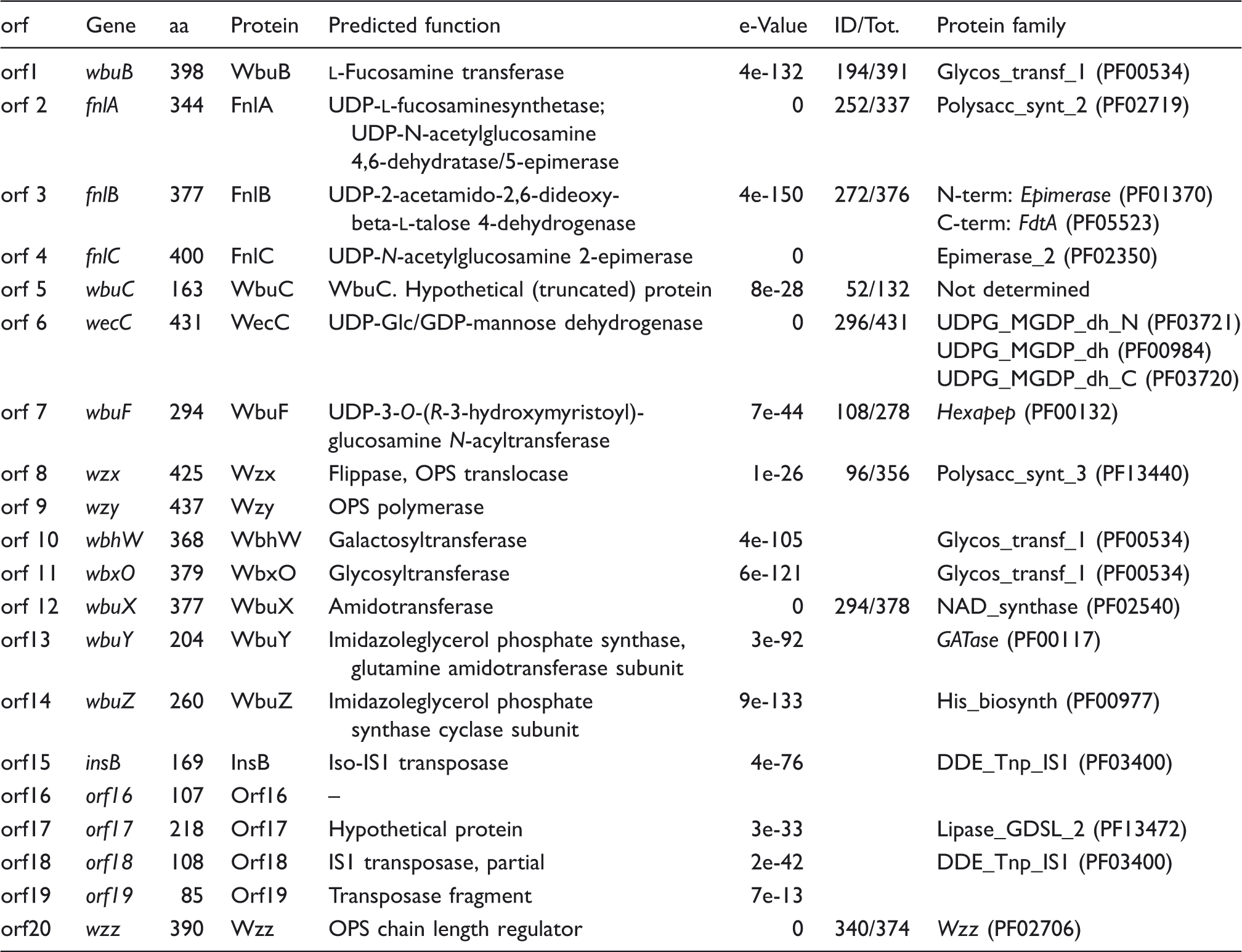
The UDP-FucpNAm genes
As the O-unit structure resembled the E. coli O145 structure having the disaccharide structure α-FucpNAm-(1 → 3)-α-GlcpNAc in common,
35
one would expect to find similarities between the biosynthetic machineries of these two organisms in the biosynthesis of the FucpNAm-precursor and the glycosyltransferase (GTase) forming the (1 → 3)-linkage to GlcNAc. Indeed, homologs for the fnlABC and wbuX genes, encoding the biosynthesis pathway to UDP-FucpNAm
35
were predicted from the gene cluster (Table 3). Orfs 2, 3, 4 and 12 showed 72, 57, 72 and 78% amino acid (aa) sequence identity to FnlA, FnlB, FnlC and WbuX of the E. coli O145 OPS gene cluster, respectively (GenBank entries AAV74535, AAV74536, AAV74537 and AAV74532). Finally, Orf1 showed 49% aa sequence identity to WbuB, the
The wbuX gene was followed by orf13 and orf14, the gene products of which showed 61 and 71% aa identity to WbuY and WbuZ of the E. coli O145 OPS gene cluster (GenBank entries AAV74533 and AAV74534). The wbuXYZ gene block is upstream of fnlA in E. coli O145, but in Y. similis O:9 it is downstream of wbuC and separated by the orf6–orf11 genes. The function of the WbuY and WbuZ proteins was predicted to be involved in providing the ammonium ion to WbuX; 35 therefore, we named these genes accordingly. Orf5 aa sequence is 43% identical to WbuC of E. coli O145 (AAV74539), a hypothetical truncated protein.
The UDP-GlcpNAcA3OAc genes
The biosynthesis pathway for 3-O-acetyl-N-acetyl-glucosaminuronic acid (UDP-GlcpNAcA3OAc) starts from UDP-GlcpNAc that is first converted to UDP-GlcpNAcA by UDP-GlcpNAc6-dehydrogenase activity. Orf6 aa sequence is 70% identical to WecC protein of Acinetobacter baumannii annotated as UDP-Glc/GDP-mannose dehydrogenase (Genbank ZP_12526711) and likely functions as the 6-dehydrogenase; therefore, it was named WecC. Orf7 remains as the sole candidate for carrying out the C-3 O-acetylation reaction as the only remaining genes orf10 and orf11 encode putative GTases (see below). Indeed, Orf7 shows highest similarity (∼ 40% aa sequence identity) to the lipid A biosynthetic enzyme UDP-3-O-(R-3-hydroxymyristoyl)-glucosamine N-acyltransferase (that carries out the C-3 O-acylation reaction); 40 therefore, Orf7 was most likely the C-3 O-acetylase, and was named as WbuF.
GTases
The GlcNAc residue is most likely the initiating sugar in the biosynthesis of the tetrasaccharide repeat unit built on the carrier lipid undecaprenyl phosphate and, consequently, WecA, the priming transferase in the biosynthesis of the enterobacterial common Ag (ECA) and wide range of OPS repeat units, 41 also functions as the priming transferase here. In bacteria, UDP-GlcNAc forms the branchpoint in the synthesis of ECA, peptidoglycan, LPS and N-acetylmannosamine, 42 and the genes needed for its biosynthesis are located outside the OPS gene cluster.
As mentioned above, WbuB is likely the FucpNAm-transferase, leaving the Galp- and GlcpNAcA3OAc-transferases to be assigned. Orfs 10 (367 aa) and 11 (379 aa) both belong to Glycos_transf_1 (PF00534) protein family. Orf10 shows 48% aa sequence identity to WbhW, a GTase of E. coli O118 and O151, and Salmonella enterica O47. These organisms have a α-
Wzx, Wzy and Wzz
These are the repeating unit processing components. 44 The O-unit synthesized on the lipid carrier is translocated across the inner membrane by a flippase, the Wzx protein, to face the periplasmic space. The flippases in different bacteria share little aa sequence similarity; however, they have 12 transmembrane domains. 45 Based on these properties Orf8 was assigned as Wzx (Table 3). Also, Wzy, the O-unit polymerase, proteins are identified mainly based on their predicted transmembrane segments; 46 based on this, Orf 9 was assigned as Wzy. Finally, Orf20 was assigned as Wzz, the polysaccharide co-polymerase that controls the chain length distribution. 47 The Wzz protein is close to 100% conserved in the Y. pseudotuberculosis complex, and this is even diagnostically relevant.48,49
Discussion
The chemical and structural studies on the LPS of Y. similis O:9 are motivated by the bacteriophage φR1-37 research.8,9,12 Phage φR1-37 was isolated in 1990 from the raw in-coming sewage of the city of Turku (Finland) using the virulence plasmid-cured, OPS-negative strainYeO3-c-R1 as a host. It was shown that out of the six sugars of the YeO3-c-R1 outer core oligosaccharide, three residues—namely Galp, GalpNAc and Sugp—furnished the structure required for phage recognition. 9 Further studies on the phage φR1-37 demonstrated that it has quite broad host range among other members of the genus Yersinia. 12 In a later, more comprehensive, host range analysis Y. similis serotype O:9 strains were also shown to be φR1-37-sensitive and that the phage φR1-37-resistant derivatives of Y. similis O:9 produce rough LPS, suggesting that the OPS functions as a phage receptor. 8 It was therefore logical to hypothesize that the Y. enterocolitica O:3 outer core and the Y. similis O:9 repeating unit share a common structural motif in their LPS that acts as bacteriophage receptor. The chemical structures of OPS are known for many serotypes of Y. pseudotuberculosis and Y. enterocolitica. The Y. pseudotuberculosis OPSs are usually branched hexosaminoglycans characterized by the presence of hexoses, 6-deoxy-hexoses, 2-acetamido-2-deoxy-hexoses and, especially, 3,6-dideoxy-hexoses.50–60 While the Y. pseudotuberculosis/Y. similis serotype O:12 OPS structure had recently been reported, 51 the composition and structure of the OPS of Y. similis O:9 was unknown.
The LPS profile of smooth strains with homopolymeric OPS such as Y. enterocolitica O:3 give a visible smear in the high molecular mass region of silver-stained gels, 2 while heteropolymeric OPSs, such as Y. enterocolitica O:8, result in a typical ladder-like banding pattern. 61 The Y. similis O:9 LPS was also smooth, with heteropolymeric-type OPS (Figure 1), as previously reported. 8 Interestingly, the bands of Y. similis O:9 LPS were present in doublets. As such doublets were not observed earlier in deoxycholate PAGE analysis 8 they are most likely artefacts of the use of SDS that might not be strong enough to fully disintegrate LPS. This is further supported by the visualization of doublet bands for the unrelated Salmonella strain analysed as a migration control. Preliminary chemical composition of complete Y. similis O:9 LPS identified various components, such as Gro, Gro-P, FucN, Ara4N, Gal, Glc, GlcNA, GlcN, Hep, Ko and Kdo. Gro and Gro-P were thought to derive from polyglycerol phosphate, an unexpected contaminant found in the LPS material by NMR spectroscopy. Hep, Ko and Kdo were known core region components, while Ara4N was often observed in lipid A.
The composition of the core region in Y. pseudotuberculosis or Y. pestis is regulated by changes in temperature, which would result in Kdo–Ko and Hep–Gal conversion.62,63 The latter conversion could also be proven for Y. similis O:9 LPS by ESI FT MS (Figure 3), as well as lipid A substitution by Ara4N (data not shown).
As Sugp was a part of the φR1-37 receptor in YeO3-c-R1, Y. similis O:9 LPS was investigated for its presence. Sugp can yield FucpN and QuiN in different proportions, depending on the reducing agents. For example, reduction with NaBH4 gives a FucpN:QuiN ratio of 7:3, while the ratio with hydrazine is 1:10. 33 However, the reduction experiments provided no evidence for the presence of Sugp in Y. similis O:9 LPS.
Methylation analysis and NMR studies (Figures 4 and 5; Table 1 and 2) generated data that helped to establish the tetrasaccharide OPS repeating unit, as depicted in Figure 6. The structure of the Y. similis O:9 OPS was branched and contained most of the previously reported components typical for closely related Y. pseudotuberculosis, namely hexoses, 6-deoxy-hexoses and 2-acetamido-2-deoxy-hexose. 64 The distinctive feature of Y. similis O:9 was the presence of hexosaminuronic acid. Unlike the other members of the Y. pseudotuberculosis complex, Y. similis O:9 OPS did not contain any of the highly immunogenic 3,6-dideoxyhexoses. 6
Phage φR1-37 receptor
Comparison of the newly elucidated OPS structure of Y. similis O:9 with the structure of the Y. enterocolitica outer core hexasaccharide23,26 revealed that Galp seemed to be the only identical residue. Thus, the epitope acting as the φR1-37 receptor could not be proposed by the simple comparison of the chemical structures. The presence of Galp in Y. similis O:9 OPS and in the Y. enterocolitica O:3 outer core oligosaccharide raised the question of whether this sugar is vital for phage recognition. Indeed, we found that the LPS of Y. similis O:9 lacking Galp in contrast to native LPS was not able to neutralize the phage, clearly suggesting that the Galp was a part of phage receptor in Y. similis O:9 OPS.
As phage receptors are also potent antigenic epitopes we wondered whether this could be elucidated by cross-reacting Abs. Thus, Ab specific for the Y. enterocolitica O:3 outer core might detect cross-reacting epitopes from the Y. similis O:9 OPS. The anti-YeO3-c-R1 antiserum reacted with Y. similis O:9 LPS, but as the cross-reactivity was visible only in the low and medium molecular mass region the most likely explanation was that the Abs recognized epitopes in the lipid A-core region, and that no cross-reacting epitopes were present between the outer core of Y. enterocolitica O:3 and the Y. similis O:9 OPS.
Supporting this conclusion was the result that mAb 2B5 did not react at all with Y. similis O:9 LPS. This indicated that the Y. similis O:9 LPS epitopes recognized by the anti-YeO3-c-R1 antiserum were not similar to Y. enterocolitica O:3 outer core. The mAb 2B5 is specific for the YeO3-c-R1 outer core hexasaccharide and the two outer core Glcp residues are important for the 2B5 epitope. 9 Absence of Glcp in the Y. similis O:9 OPS might partly explain the lack of reactivity with mAb 2B5.To elucidate the phage receptor in Y. similis O:9 further studies are needed. It is possible that both the outer core region and OPS share common conformational properties resulting in receptor formation by appropriately-oriented side groups in the sugars and not by identical sugar residues. Another, more plausible, explanation is that the phage expresses several receptor binding proteins allowing it to bind to several different LPS structures. Supporting this possibility is the fact that φR1-37 can also infect Y. enterocolitica serotype O:50, the structure of which was recently established and also the O:50 OPS shows no similarity with the Y. enterocolitica O:3 outer core. 65
The comparison of the UDP-FucNAm biosynthesis and GTase gene organizations of the Y. similis O:9 and E. coli O145 gene clusters revealed that the fnlABC gene block is conserved; however, the locations of the wbuB and wbuXYZ genes are not (Figure 8). The wbuB gene has moved from between fnlC and wbuC in front of fnlA. It is also remarkable that the wbuC gene is conserved, as it was predicted to be a gene remnant in E. coli O145 and O26 originating from waaK of E. coli K-12.35,66 Remarkably, this gene is present in the truncated form in many organisms, and a simple BLAST search returned over 100 significant hits. Provided the gene is just a non-functional remnant, one would expect limited sequence conservation of the aa sequences; therefore, we predict that the gene is still functional. However, further work is needed to elucidate this.
Footnotes
Funding
The research presented in this article was partly funded by the Academy of Finland (project 1114075 to M.S.).
Acknowledgements
We thank Dr. Buko Lindner for help with ESI-MS; Hermann Moll and Regina Engel for help with GC–MS; Heiko Käßner for NMR recordings; and Dr. Katarzyna Anna Duda and Dr. Katarzyna Kasperkiewicz for providing YeO3-c-R1 antisera.