
Abstract
Endotoxemia is a major cause of chronic inflammation, and is an important pathogenic factor in the development of metabolic syndrome and atherosclerosis. Human apolipoprotein E (apoE) and apoA-I are protein components of high-density lipoprotein, which have strong anti-endotoxin activity. Here, we compared anti-endotoxin activity of Ac-hE18A-NH2 and 4F peptides, modified from model amphipathic helical 18A peptide, to mimic, respectively, apoE and apoA-I properties. Ac-hE18A-NH2, stronger than 4F, inhibited endotoxin activity and disaggregated Escherichia coli 055:B5 (wild smooth serotype). Ac-hE18A-NH2 and 4F inhibited endotoxin activity of E. coli 026:B6 (rough-like serotype) to a similar degree. This suggests that Ac-hE18A-NH2 as a dual-domain molecule might interact with both the lipid A and headgroup of smooth LPS, whereas 4F binds lipid A. In C57BL/6 mice, Ac-hE18A-NH2 was superior to 4F in inhibiting the inflammatory responses mediated by E. coli 055:B5, but not E. coli 026:B6. However, in THP-1 cells, isolated human primary leukocytes, and whole human blood, Ac-hE18A-NH2 reduced responses more strongly than 4F to both E. coli serotypes either when peptides were pre-incubated or co-incubated with LPS, indicating that Ac-hE18A-NH2 also has strong anti-inflammatory effects independent of endotoxin-neutralizing properties. In conclusion, Ac-hE18A-NH2 is more effective than 4F in inhibiting LPS-mediated inflammation, which opens prospective clinical applications for Ac-hE18A-NH2.
Introduction
Inflammation in humans due to infection is often associated with Gram-negative bacteria. LPS, an endotoxin found on the outer membrane of Gram-negative bacteria, triggers an innate immune response that is characterized by production of pro-inflammatory cytokines and reactive oxygen species.1–3 TNF-α and IL-6 are important mediators of an LPS-activated inflammatory cascade leading to injuries to blood vessels and organ tissues.1,4 A number of experimental and clinical studies suggest that LPS-mediated endotoxemia promotes atherosclerosis.5–15 TLR4, the major cellular target for LPS in humans, may play a role in the initiation and progression of inflammation and atherosclerosis. 10 In response to endotoxin, increased adhesion of monocytes and T cells to aortic endothelium is found in a rodent model. 9 It has also been demonstrated that LPS promotes the binding of oxidized low-density lipoprotein (LDL) to activated human macrophages resulting in foam cell formation. 5 Pro-atherogenic activation of macrophages via TLR4 can occur cooperatively by low subclinical concentrations of endotoxin and minimally oxidized LDL. 6 Higher levels of LPS in plasma and/or in arterial wall are shown to be associated with increased carotid atherosclerosis.7,8,11 A link has been demonstrated between periodontal disease-associated endotoxemia and atherosclerosis.12–15 In addition, LPS-mediated inflammation may contribute to metabolic disorders in humans.16,17
Plasma lipoproteins play an important role in neutralization of LPS.18–21 Apolipoproteins are the protein components of lipoproteins. Among various apolipoproteins, apolipoprotein A-I (apoA-I) and apolipoprotein E (apoE) appear to possess anti-inflammatory and anti-atherogenic properties.22–24 In many laboratories, different apoA-I and apoE mimetic peptides have been synthesized and tested in different animal models for their anti-inflammatory and anti-atherogenic efficacy.25–28 In our laboratory, we designed peptides that mimic apoA-I and apoE properties, and demonstrated that they exert strong anti-atherogenic effects in animal models of atherosclerosis.29–34 One of these peptides, 4F, was derived from the class A amphipathic helix peptide 18A.35–37 As shown in Figure 1, peptide 18A is only 18 amino acid residues in length (compared to 243 amino acids present in human apoA-I). It has no sequence similarity with apoA-I, but it mimics the class A amphipathic helixes contained in apoA-I and some apoA-I lipid-binding and anti-inflammatory properties.35,37 It has been reported that administration of the apoA-I mimetic peptide 18A prolongs survival in LPS-injected mice.
38
To improve apoA-I-mimicking properties, 18A peptide has been modified to 4F by substitutions of two Leu to Phe, which increased hydrophobicity and, thus, lipid affinity of the resulting peptide (Figure 1).
39
4F mimics apoA-I anti-atherogenic properties.
40
Though 4F does not decrease plasma cholesterol, it exerts strong anti-inflammatory properties.30,41 In different in vitro and in vivo experiments, 4F demonstrated a high efficacy in inhibiting LPS-mediated inflammation.42–44 Direct LPS binding and neutralization, similar to that of HDL/apoA-I, has been suggested to be one of the important mechanisms of anti-endotoxin activity of 4F.
42
In recent studies in selected patient groups with high-risk cardiovascular disease, acceptable safety profiles in humans have been reported for parenteral and oral forms of 4F.45,46
A schematic of modification of model class A amphipathic α-helical peptide 18A to 4F and Ac-hE18A-NH2. Peptides are presented as a helical net. The amino acids are shown in one-letter code. The hydrophobic residues are in bold circles, and the charges of the amino acids are also shown. To improve apoA-I-mimicking properties, 18A peptide has been modified to 4F by substitution of two Leu to Phe. This increased hydrophobicity and, thus, lipid affinity of 4F. In another modification, covalently linking LRKLRKRLLR (141–150 residues from receptor-binding region of apoE) to 18A produced Ac-hE18A-NH2, which mimics apoE effects. This modification also increased hydrophobicity and increased positive charge to the resulting peptide.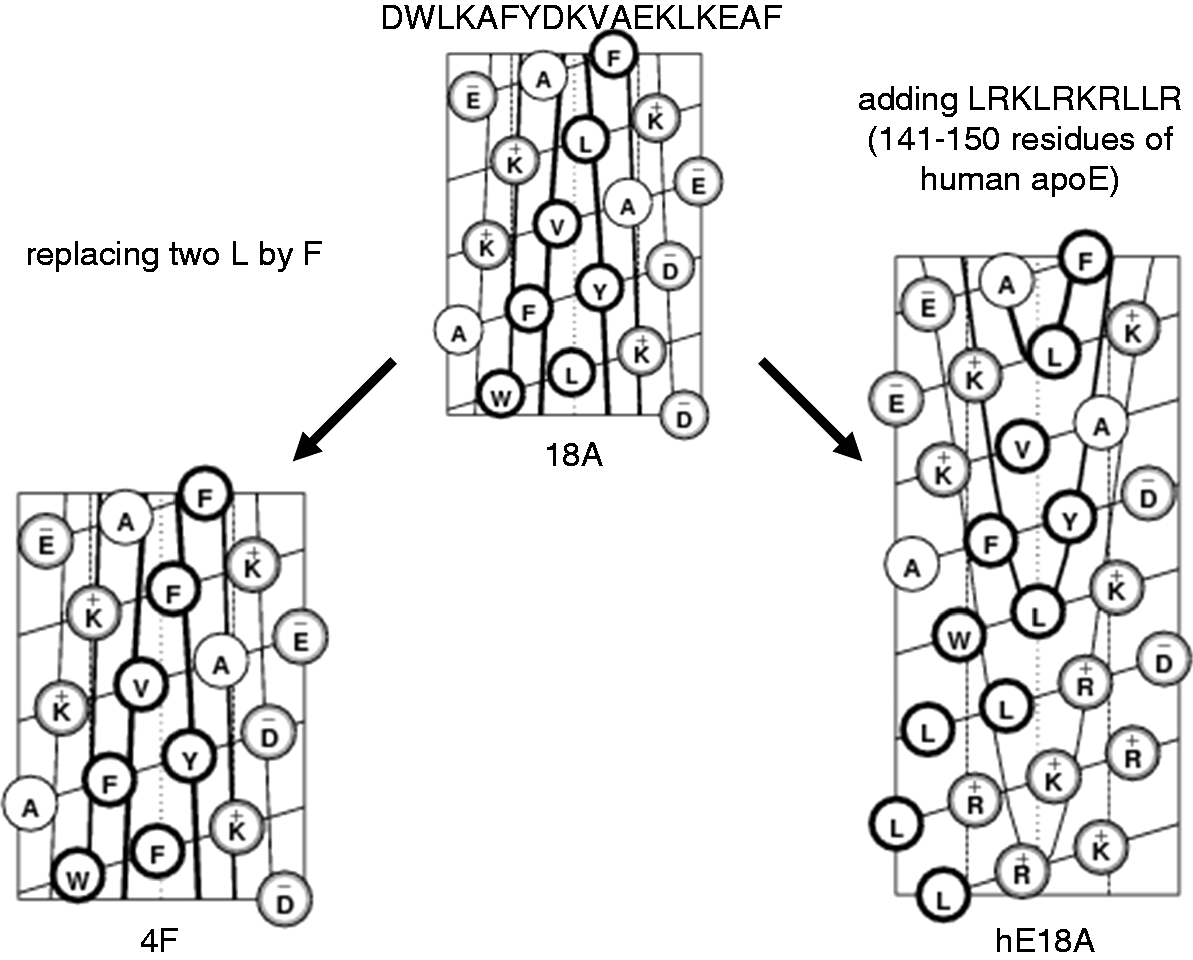
Covalently linking LRKLRKRLLR (residues 141–150 from the receptor-binding region of apoE) 27 to 18A peptide with lipid-binding properties 37 produced Ac-hE18A-NH2 (Figure 1), which mimics apoE effects and reduces plasma cholesterol in several animal models.27,31,47,48 In THP-1 monocyte-derived macrophages and HepG2 cells, Ac-hE18A-NH2 demonstrated recycling properties, which increased formation of preβ-HDL, 49 analogous to apoE recycling in hepatocytes and macrophages. 50 Previously, strong anti-inflammatory activity of human apoE against LPS-mediated inflammation has been shown in rodents.51–53 A direct anti-viral and Gram-negative microbial activity of the receptor-binding region of human apoE has been reported. 54 While each of the Ac-hE18A-NH2 domains independently exerts anti-inflammatory properties, combining these domains together does not necessary lead to amplification of such anti-inflammatory properties. In a recent report, Ac-hE18A-NH2 inhibited LPS-mediated inflammatory response in THP-1 monocyte-derived macrophages and HUVEC cells. 49 Here, we sought to test whether Ac-hE18A-NH2 produces efficient anti-endotoxin activity in vitro and in vivo in comparison to 4F, which has well-described anti-endotoxin activity.42–44,55 Two serotypes of LPS, Escherichia coli 055:B5 and E. coli 026:B6, were mostly used in the present work. While E. coli 055:B5 is a typical smooth serotype of E. coli with long O-chain, E. coli 026:B6 is rather atypical E. coli serotype with a short O-chain, resembling rough mutant LPS strain.
Here we demonstrate that Ac-hE18A-NH2, analogous to 4F, interacts with LPS and neutralizes endotoxin activity both in vitro and in vivo. In THP-1 cells, human blood in vitro and C57BL/6 mice, Ac-hE18A-NH2 exerts stronger post-LPS anti-inflammatory effects than 4F, especially when LPS with a longer O-chain than E. coli 055:B5 was used.
Materials and methods
Peptides and LPS
Ac-hE18A-NH2 (Ac-L-R-K-L-R-K-R-L-L-R-D-W-L-K-A-F-Y-D-K-V-A-E-K-L-K-E-A-F-NH2) and L-4F (Ac-D-W-F-K-A-F-Y-D-K-V-A-E-K-F-K-E-A-F-NH2) were synthesized by the solid phase peptide synthesis method using fluorenylmethyloxycarbonyl amino acids and suitable protected amino acids as described previously.39,47 LPS was obtained commercially from Sigma Aldrich (St Louis, MI, USA), (E. coli 026:B6, E. coli 011:B4, E. coli 055:B5 and Pseudomonas aeruginosa 10). Bodipy–LPS (E. coli 055:B5; Molecular Probes; Eugene, OR, USA) was purchased from Invitrogen (Carlsbad, CA, USA).
Measurements of peptide effects on endotoxin activity
Endotoxin activity of LPS in aqueous solutions (saline) and in mouse plasma in the absence and presence of peptides was measured by limulus amoebocyte lysate (LAL) assay using kinetic-chromogenic test (Endochrome-K; Charles River; Charleston, SC, USA) or kinetic-turbidimetric test (KTA2; Charles River; Charleston SC, USA).
In vivo experiments with LPS-mediated inflammation in mice
C57BL/6 female mice of 14–16-wk-old (Jackson Laboratory, Bar Harbor, ME, USA) were used throughout the study. Prior to experiments, the mice were acclimatized for 1 wk in a 12 h light/dark cycle with free access for food and water. Non-fasted mice underwent either injection of LPS (E. coli 026:B6, 25 µg, i.p.) or saline (100 µl) followed by the administration of Ac-hE18A-NH2 (100 µg, i.v., retro-orbitally) or L-4F (100 µg, i.v.) or saline (100 µl) after 5 min. Blood samples were collected at 3 h and at 27 h (at 24 h, mice were fasted) post-LPS. The concentrations of IL-6 and serum amyloid A (SAA) in mouse plasma were quantified using commercially obtained ELISA kits (BD Biosciences; San Jose, CA, USA; and Tridelta Development, Maynooth, Ireland, respectively). Relative changes in reactive oxygen species (ROS) levels in plasma were measured using a 2′,7′-dichlorofluorescin (DCF; Sigma-Aldrich; St Louis, MI, USA) assay. Plasma (5 µl) was diluted in 200 µl of PBS containing 50 µM DCFH and incubated in 96-well black plates for 1 h at 37℃ in the dark. Fluorescence was then measured with excitation and emission wavelengths of 485 nm and 530 nm, respectively, and then normalized to total plasma cholesterol. Plasma triglycerides and total cholesterol were measured using corresponding commercial kits (Thermo Scientific; Middletown, VA, USA). In another set of experiments, Ac-hE18A-NH2 was administered 30 min pre- or 3 h post-LPS, and its effects on plasma cholesterol, triglycerides and relative ROS changes were measured as indicated. In the final experiment, we compared effects of both peptides (100 µg, i.v.) administered 5 min after a different LPS (E. coli 055:B5, 50 µg, i.p.). We measured plasma levels of IFN-γ in blood samples collected at 3 h and 24 h post-LPS using a commercially obtained ELISA kit (BD Biosciences; San Jose, CA, USA). At 3 h post-LPS, we also measured plasma endotoxin activity as described above.
Experiments with LPS and Bodipy-LPS in saline
To measure a direct effect of Ac-hE18A-NH2 or L-4F on endotoxin activity, we mixed LPS from different species with peptide in saline and immediately measured endotoxin activity as described above. To demonstrate a direct binding of peptides with LPS, peptide and LPS of indicated concentrations were incubated in saline and then subjected to native agarose gel electrophoresis (0.75% Tris/Tricine gel, at 130V). Changes of electrophoretic mobility of peptides were assessed by gel staining with GelCode Blue Stain Reagent for proteins (Pierce; Rockford, IL, USA). Effects of Ac-hE18A-NH2 and L-4F on Bodipy-LPS aggregation in saline were examined by measuring fluorescent emission at 530 nm in eluent fractions separated by size exclusion chromatography. Concentrations of LPS and peptides are specified in the text and figure legends.
In vitro experiments with human blood
Blood samples from healthy volunteers were collected by venipuncture into heparinized Vacutainer tubes (BD Biosciences; San Jose, CA, USA). To 1 ml of blood (1) saline; (2) L-4F (40 µg/ml); (3) Ac-hE18A-NH2 (40 µg/ml); (4) LPS (1 µg/ml, E. coli 026:B6); (5) LPS and L-4F; or (6) LPS and Ac-hE18A-NH2 was added and incubated for 6 h at 37℃ in 5% CO2. Plasma was isolated by centrifugation, and concentration of TNF-α and IL-6 was quantified by using commercially obtained ELISA kits (BD Biosciences; San Jose, CA, USA) specific for humans.
In vitro measurement with isolated human leukocytes
Fresh human leukocytes were collected from the blood of healthy volunteers by aspiration of a buffy coat. Any remaining red blood cells in buffy coat were lysed; leukocytes were then centrifuged and washed twice in PBS. Leukocytes (1–2 × 106 cells/ml) were then re-suspended in 50% donor plasma/50% PBS and stimulated with 1 µg/ml LPS (E. coli 026:B6) in the absence and presence of 40 µg/ml L-4F or Ac-hE18A-NH2. Lucigenin (10 µM) was added 2 min before adding LPS and/or peptides and onset of measurements. Lucigenin-amplified chemiluminescence was measured for 3 h as a marker of superoxide formation. 56 Area under curves (AUC) of relative photon emission (RPE) over time was calculated.
In vitro experiments with human monocyte THP-1 cells
THP-1 cells (ATCC, Manassas, VA, USA) were maintained in complete RPMI 1640 medium (ATCC) supplemented with 10% heat-inactivated FBS, 2 mM
Study approval
All human studies were approved by the University of Alabama at Birmingham (UAB) institutional review board. All healthy volunteers provided written informed consent. All procedures involving mice were conducted in accordance with the guidelines of the Care and Use of Laboratory Animals and the National Institutes of Health and approved by the Institutional Animal Care and Use Committee of the UAB.
Statistical analysis
All results, unless otherwise specified, are reported as the mean ± SEM. Statistical analysis was performed using GraphPad Prism V.4.0.1. Differences between groups were assessed by paired or unpaired one-way ANOVA with post-hoc Bonferroni's Multiple Comparison test. A value of P < 0.05 was considered to be statistically significant.
Results
Effects on endotoxin activity and LPS-mediated cytokine production in human blood in vitro
Fresh human blood from healthy volunteers was stimulated by 1 µg/ml LPS (E. coli O26:B6) with and without Ac-hE18A-NH2 or L-4F (both 40 µg/ml) for 6 h at 37℃ and 5% CO2 (n = 4). Highly elevated plasma endotoxin activity was measured in LPS-stimulated samples using the LAL test (P < 0.05 versus control; Figure 2A), which was associated with increased plasma levels of TNF-α and IL-6 (P < 0.05 versus control; Figure 2B, C). Peptides significantly decreased plasma endotoxin activity in LPS-stimulated samples, as well as plasma TNF-α and IL-6 levels (Figure 2A–C). Ac-hE18A-NH2 was superior to L-4F in inhibiting LPS-induced IL-6 production in human blood (P < 0.05, Figure 2C).
Ac-hE18A-NH2 and L-4F inhibit inflammatory response to LPS in isolated human blood and in primary human leukocytes. (A–C) Fresh human blood from healthy volunteers was stimulated by 1 µg/ml LPS (E. coli O26:B6) with and without Ac-hE18A-NH2 or L-4F (both, 40 µg/ml). Blood was incubated in a sterile incubator for 6 h at 37℃ in 5% CO2 (n = 4). Plasma endotoxin activity (A), plasma levels of TNF-α (B) and IL-6 (C) were measured. (D, E) Freshly isolated human leukocytes were incubated in 50% donor plasma/50% PBS in the presence of 1 µg/ml LPS (E. coli O26:B6) with and without Ac-hE18A-NH2 or L-4F (both, 40 µg/ml). LPS-induced superoxide formation was measured by lucigenin (10 µM)-mediated chemiluminescence at 37℃ in air (n = 5). A representative experiment performed in triplicate (D) and the calculated AUC from a total of five experiments (E) are shown. *P < 0.05, **P < 0.01 versus LPS, †P < 0.05 versus controls, ‡P < 0.05 versus LPS plus L-4F.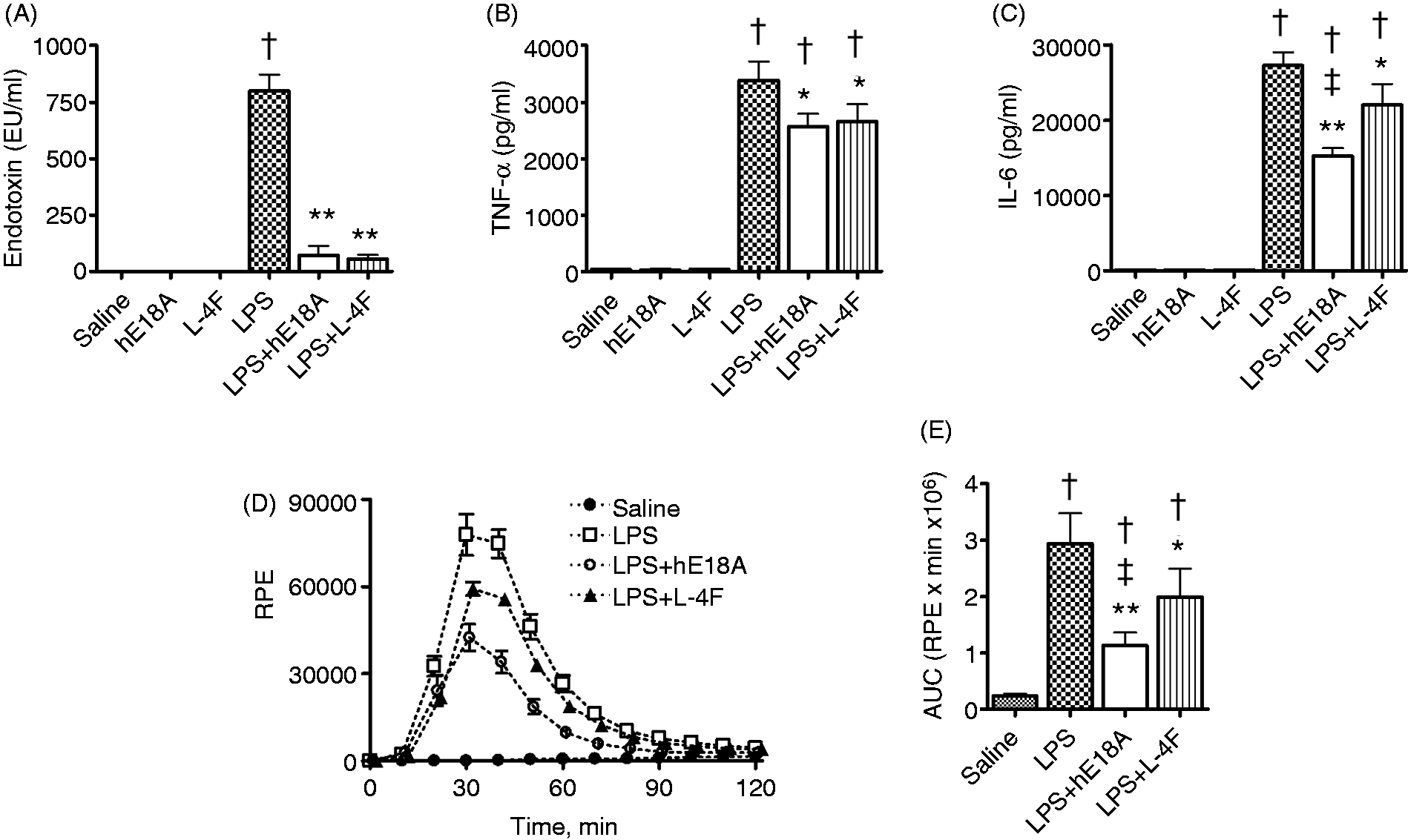
Effects on LPS-mediated superoxide production in primary human leukocytes
Freshly isolated human leukocytes were incubated in healthy donor plasma with 1 µg/ml LPS (E. coli O26:B6) and/or Ac-hE18A-NH2 or L-4F (40 µg/ml). Superoxide formation was measured using lucigenin-amplified chemiluminescence. Whereas Ac-hE18A-NH2 or L-4F alone did not cause any chemiluminescence in the samples (not shown), LPS alone resulted in enhanced chemiluminescence (P < 0.001 versus saline; Figure 2D, E). Both Ac-hE18A-NH2 and L-4F significantly inhibited the rate, as well as peak, of LPS-mediated chemilumescence emission (P < 0.001 for Ac-hE18A-NH2 and P < 0.05 for L-4F versus LPS), with Ac-hE18A-NH2 having a greater inhibition than that of L-4F (P < 0.05; Figure 2D, E).
Effects on cytokine production induced by different LPS species in human THP-1 cell line
Human THP-1 cells (106 cells in 1 ml) were stimulated with LPS (0.5 µg/ml) from E. coli (O55:B5, O26:B6, and O11:B4) and P. aeruginosa in the presence or absence of peptides (10 µg/ml) (n = 4). Incubation for 22 h with LPS alone resulted in significant elevation of TNF-α and IL-6 levels in cell media (Figure 3). Ac-hE18A-NH2 significantly inhibited TNF-α and IL-6 release in cell media (P < 0.01 versus LPS), whereas L-4F has no effect on TNF-α release and inhibited only IL-6 release in cells stimulated by E. coli O26:B6 and O114:B4 (P < 0.05 versus LPS). In all treatment groups, Ac-hE18A-NH2 inhibited LPS-mediated cytokine production more effectively than L-4F (P < 0.05; Figure 3). In a similar, separate experiment, a much higher concentration of L-4F (50 µg/ml) was used to significantly inhibit TNF-α release by LPS-stimulated THP-1 cells (not shown).
Ac-hE18A-NH2 is superior to L-4F in inhibiting inflammatory response to LPS in human monocyte THP-1 cell line. Various LPS types (as specified in the figure, 0.5 µg/ml) were used to stimulate 106 THP-1 cells in 1 ml for 22 h at 37℃ and 5% CO2 (n = 4). Peptides (10 µg/ml) were added immediately after LPS. TNF-α and IL-6 concentrations in cell media were measured by ELISA. *P < 0.05, **P < 0.01, ***P < 0.001 versus LPS, †P < 0.05 versus controls, ‡P < 0.05 and ‡‡P < 0.01 versus LPS plus L-4F. hE18A: Ac-hE18A-NH2.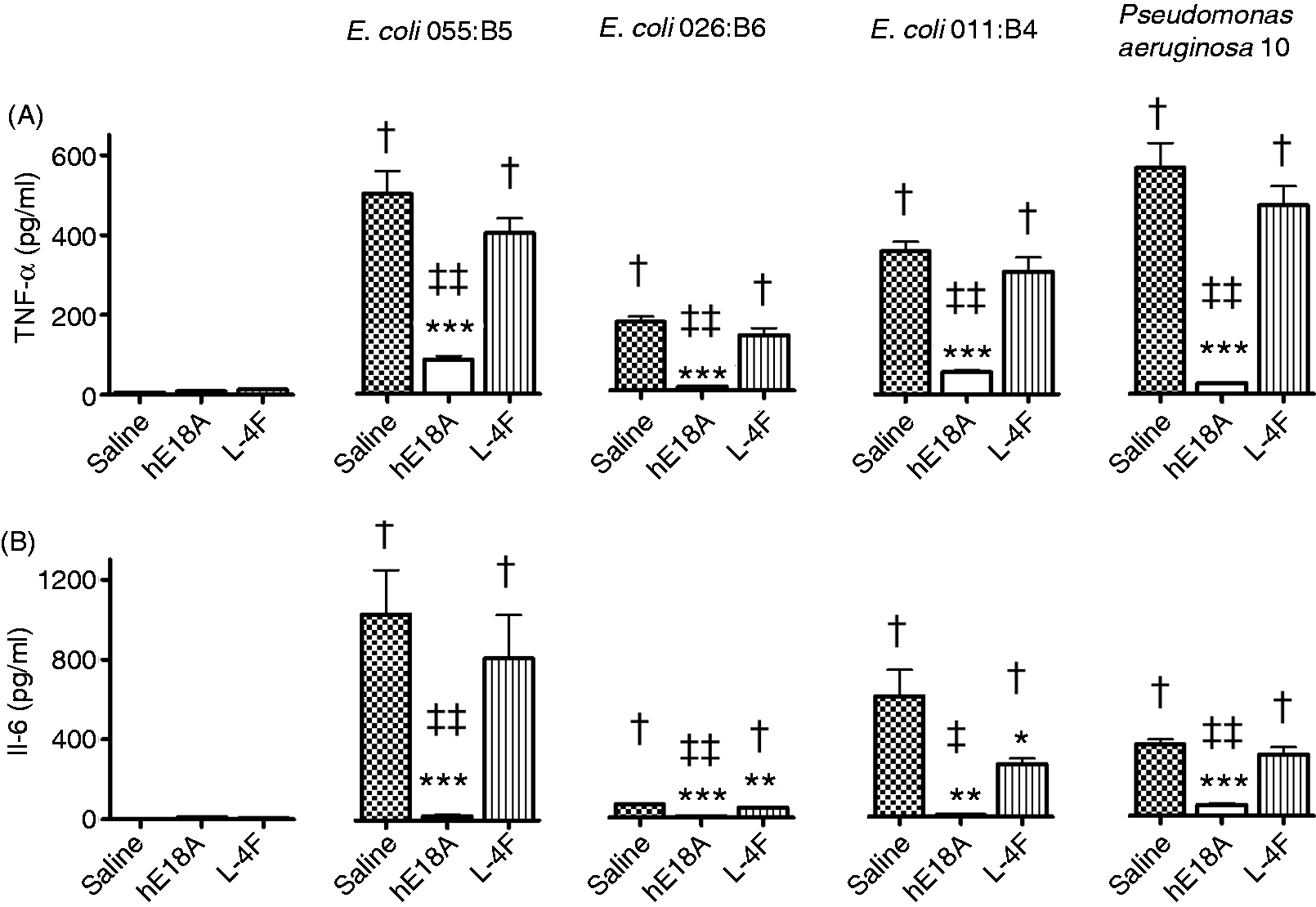
Effects on endotoxin activity in aqueous solution
We compared neutralizing activity of Ac-hE18A-NH2 and L-4F against various LPS [E. coli 055:B5 (0.5 µg/ml), E. coli 026:B6 (0.5 µg/ml) and P. aeruginosa 10 (1 µg/ml)]. LPS was mixed with Ac-hE18A-NH2 or L-4F at peptide doses of 0, 1, 10 and 100 µg/ml in saline, and endotoxin activity was immediately measured using LAL assay. As presented in Figure 4, both peptides neutralized the endotoxin activity of E. coli 055:B5 and E. coli 026:B6 in a dose-dependent manner (P < 0.001). Ac-hE18A-NH2 inhibited endotoxin activity of E. coli 055:B5 more effectively than L-4F (P < 0.05, n = 6; Figure 4A). Both peptides similarly inhibited endotoxin activity of E. coli 026:B6 (not significant, n = 5; Figure 4B). In a separate experiment, Ac-hE18A-NH2 and L-4F also demonstrated strong endotoxin neutralizing activity against P. aeruginosa 10 (not shown).
Ac-hE18A-NH2 and L-4F inhibit endotoxin activity in aqueous solution. LPS from E. coli 055:B5 (0.5μg/ml, n = 6) (A) and from E. coli 026:B6 (0.5μg/ml, n = 5) (B) was mixed with Ac-hE18A-NH2 or L-4F at the indicated concentrations in saline and endotoxin activity was immediately measured with the LAL assay. ‡P < 0.05 versus LPS plus L-4F. hE18A: Ac-hE18A-NH2.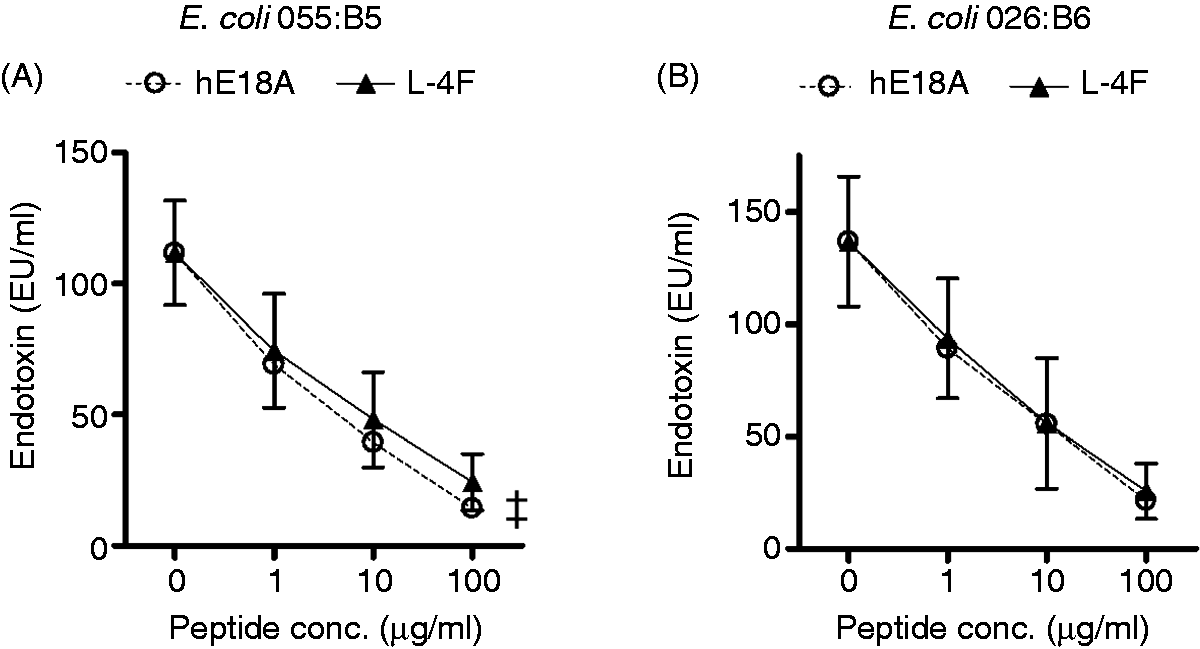
Binding with LPS in aqueous solution
Neutralization of endotoxin activity in aqueous solution suggests a direct interaction of peptides with LPS. L-4F binding to LPS has been previously demonstrated.42,55 Here, we demonstrate the binding of Ac-hE18A-NH2 to LPS in comparison to that of L-4F. Each peptide (1 µg) was mixed to increasing doses of LPS E. coli 055:B5 (0, 1, 5, 10 µg), and the mixture was separated in agarose gel electrophoresis (Figure 5A). Peptides without LPS moved to cathode end of the gel, which is observed for positively charges molecules. Interestingly, in the presence of 1 µg of LPS, less peptides is seen in the cathode side of the gel. Moreover, at 5 and 10 µg of LPS, all peptides moved to anode side of the gel. This phenomenon was consistently observed in six similar experiments. It is explained by binding of peptides to negatively charged LPS molecules, with overall net negative charge of such complex. When a similar experiment was repeated with E. coli 026:B6 serotype, which has a typically short O-chain, no, or a very little amount of, peptide was detected at anode side of the gel (not shown). The lack of mobility might reflect mutual neutralization of positively charged peptides and negatively charged LPS core.
Ac-hE18A-NH2 and L-4F bind and disaggregate LPS. (A) Each peptide (1μg) was mixed with increasing concentrations of LPS E. coli 055:B5 (0, 1, 5, 10 µg), and the mixture was separated by agarose gel electrophoresis (n = 6). A representative gel is shown. (B) Disaggregation of Bodipy–LPS (5 µg/ml E. coli O55:B5, B-LPS) by Ac-hE18A-NH2 (100 µg/ml) and L-4F (100 µg/ml, inset). After incubation of Ac-hE18A-NH2 (n = 4) or L-4F (n = 5) with Bodipy–LPS in saline, fluorescent emission of eluent fractions separated by size exclusion chromatography was measured at 530 nm. Representative examples are shown. (C) LPS presence in selected FPLC eluents was also detected by measuring LAL endotoxin activity. Peaks of Bodipy fluorescence in eluents co-localize with peaks of LAL endotoxin activity. hE18A: Ac-hE18A-NH2.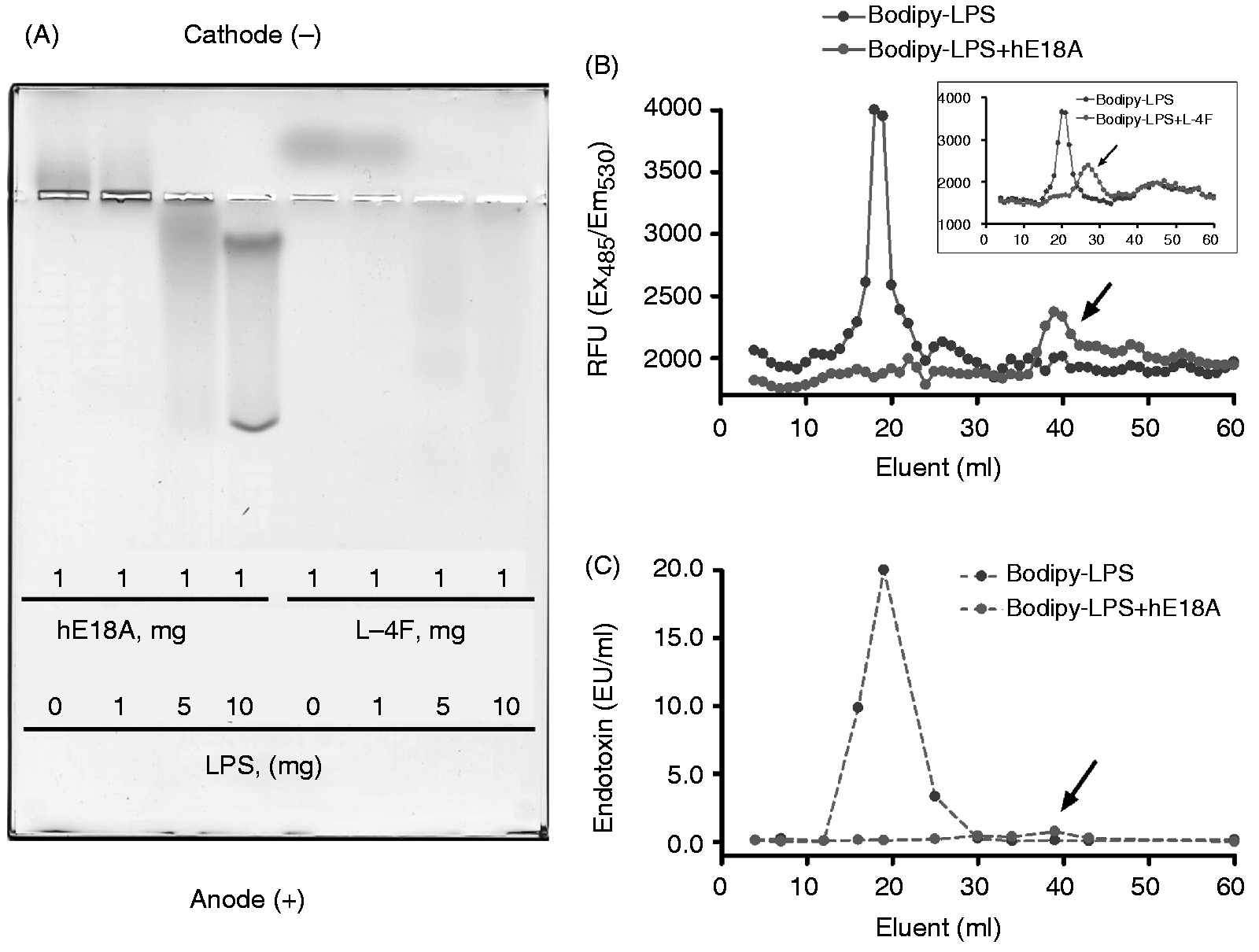
Effects on LPS aggregation in aqueous solution
A number of studies suggest that LPS in the aggregated state is biologically active.58,59 Therefore, to evaluate the binding activity of Ac-hE18A-NH2 and its effect on the aggregation state of LPS in comparison to L-4F, 42 we incubated Bodipy-LPS (5 µg/ml E. coli O55:B5) in PBS with and without Ac-hE18A-NH2 or L-4F (100 µg/ml) for 20 min, and measured Bodipy fluorescence in fast protein liquid chromatography (FPLC) eluents (Figure 5B). Without peptides, a large peak was observed in the earlier fractions (around 20-ml eluents) indicating Bodipy–LPS aggregates. Ac-hE18A-NH2 disaggregated these Bodipy–LPS aggregates and produced Ac-hE18A-NH2–Bodipy–LPS complexes of smaller size (around 40-ml eluents) as revealed by gel filtration chromatography. L-4F also produces disaggregation of large LPS aggregates, but to a lesser extent than Ac-hE18A-NH2 (around 25–30-ml eluents, as shown in the inset in Figure 5B). We traced LPS presence by measuring LAL endotoxin activity in selected FPLC eluents to confirm that the disappearance of the large fluorescent peak in the presence of Ac-hE18A-NH2 reflects actual translocation of Bodipy-LPS (Figure 5C).
Impact of serum on peptide effects in human THP-1 cells
To evaluate a role of serum in the mechanisms of anti-inflammatory activity of the peptides against LPS, we compared the production of TNF-α and IL-6 by THP-1 cells, which were incubated either in media containing 0.2% BSA (serum-free media) or 10% FBS, and stimulated by 0.5 µg/ml LPS (E. coli 055:B5). Consistent with earlier reports that LPS similarly stimulates the production of TNF-α by THP-1 cells in serum and serum-free media,
57
we also found similar levels of LPS-induced TNF-α in both THP-1 cell medias following 20 h incubation with LPS (827 ± 68 pg/ml in 10% PBS and 978 ± 102 pg/ml in 0.2% BSA; not significant, n = 4). L-4F (10 µg/ml) inhibited TNF-α production in both cell medias [by 25% in 10% FBS (P < 0.05 versus LPS) and by 33% in 0.2% BSA (P < 0.01 versus LPS)]. Though the inhibitory effect of L-4F was somewhat stronger in serum-free media than in serum, the difference was not statistically significant (Figure 6A). In contrast, Ac-hE18A-NH2 (5 µg/ml) inhibited TNF-α production much more strongly in serum-free media than in media with 10% PBS (by 90% versus 66%, respectively; P < 0.05). In either media, the effects of Ac-hE18A-NH2 were superior to that of L-4F (P < 0.001; Figure 6A). LPS alone also caused elevation of IL-6 in both cell media (1916 ± 231 pg/ml in 10% FBS and 1706 ± 141 pg/ml in 0.2% BSA; not significant). As in our previous experiment, Ac-hE18A-NH2 significantly inhibited IL-6 elevation, whereas L-4F did not (Figure 6B). Inhibition of LPS-induced IL-6 elevation by Ac-hE18A-NH2 was similar in cell media with 10% FBS and 0.2% BSA (Figure 6B).
Serum plays little or no role on peptide effects in human THP-1 cells. A role of serum in the mechanisms of anti-inflammatory activity of the peptides against LPS was studied by measuring the production of TNF-α (A) and IL-6 (B) by THP-1 cells, which were incubated either in media containing 0.2% BSA (serum-free media) or 10% FBS and stimulated by 0.5 µg/ml LPS (E. coli 055:B5, n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus LPS, †P < 0.05 versus controls, ‡‡‡P < 0.001 versus LPS plus L-4F. hE18A: Ac-hE18A-NH2.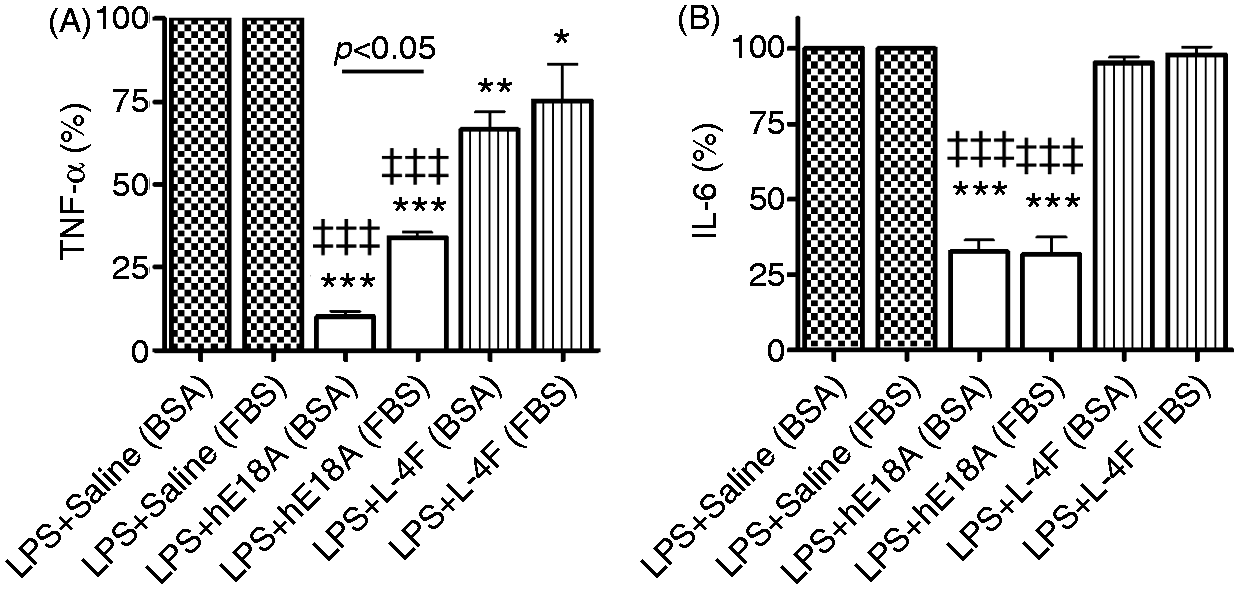
Impact of cell- and LPS-dependent mechanisms in inhibition of LPS-induced response in THP-1 cells
In previous experiments, direct anti-inflammatory effects of peptides on cells, which are different from LPS-neutralizing effects, have been suggested.49,60 Here, we conducted parallel experiments on THP-1 cells to (1) compare the protective effects of peptides used to treat cells for 20 h with complete peptide removal before cell stimulation with LPS for 20 h; and (2) to compare the protective effects of pre-incubation with peptides versus co-incubation with peptides against LPS stimulation. When THP-1 cells were pretreated with saline alone, LPS (0.5 µg/ml, E. coli 055:B5) resulted in high elevation of TNF-α levels in cell media (P < 0.001 versus control; Figure 7A). Pretreatment of cells with Ac-hE18A-NH2 (5 µg/ml) inhibited LPS effects by 30% (P < 0.01), whereas pretreatment with L-4F (10 µg/ml) had little inhibitory effect on cell response to LPS (by 14%, not significant). When co-incubated with LPS, Ac-hE18A-NH2 and L-4F both strongly inhibited cell responses to LPS (P < 0.001 for Ac-hE18A-NH2 and P < 0.05 for L-4F versus LPS). The protective effect of peptides during co-incubation with LPS was approximately twice as efficient than that of during pre-incubation (Figure 7A). Similar results were obtained in the experiments with E. coli 026:B6 (Figure 7B).
Ac-hE18A-NH2 but not L-4F, produces strong cell-dependent effects in addition to LPS-dependent mechanisms in inhibition of LPS-induced response in THP-1 cells. (A) Experiments with E. coli 055:B5 (n = 4); (B) Experiments with E. coli 026:B6 (n = 4). ‘LPS after peptide’ = THP-1 cells were incubated with peptide for 20 h as a pretreatment, followed by thorough washing of peptide from cells and incubation with LPS (0.5 µg/ml) for 20 h. ‘LPS+peptide’ = THP-1 cells were pretreated with saline for 20 h, and washed and incubated with LPS and peptide for 20 h. *P < 0.05, **P < 0.01, ***P < 0.001 versus LPS, †P < 0.05 versus controls, ‡‡P < 0.01 versus LPS plus L-4F. hE18A: Ac-hE18A-NH2.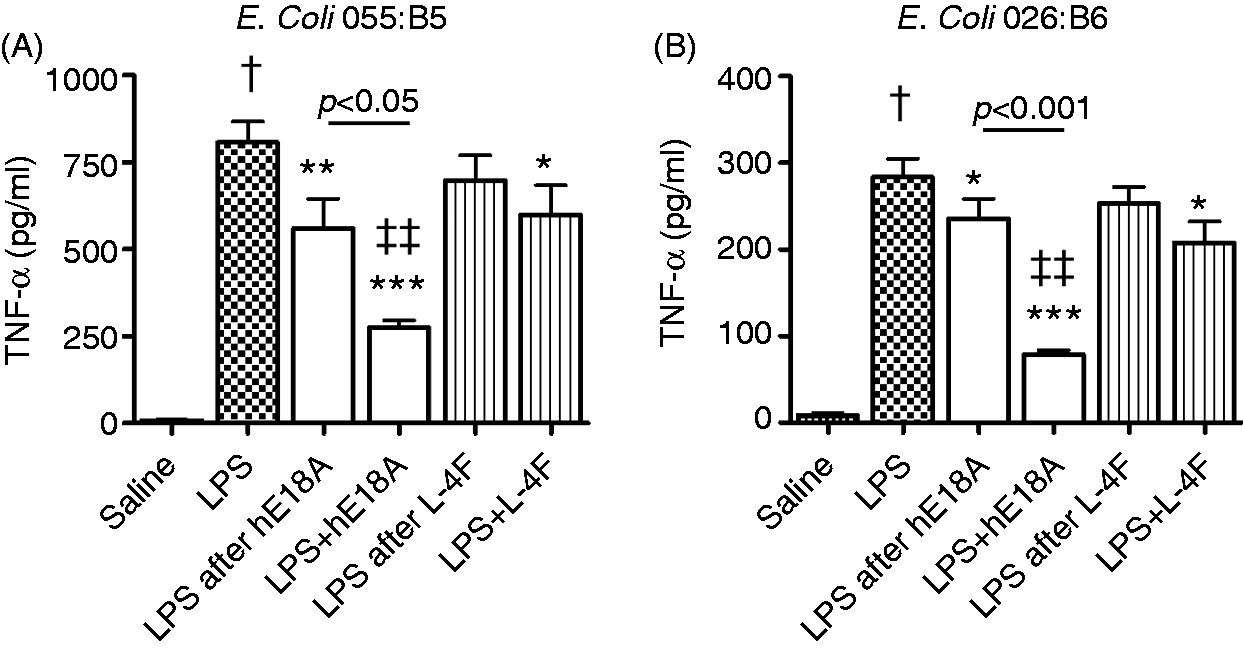
Effects on endotoxin activity and LPS-mediated inflammatory response in mice
Either peptides (100 µg, i.v.) or saline (100 µl) were administered 5 min after LPS injection (E. coli O26:B6, 50 µg, i.p.) in C57BL/6 mice. At 3 h post LPS, saline-treated mice had high levels of plasma endotoxin activity (P < 0.05 versus control mice), which was significantly reduced by peptides (P < 0.05 versus LPS; Figure 8A). Plasma levels of IL-6 and relative levels of ROS were highly elevated after LPS in saline-treated mice compared with control mice (Figure 8B, C). Both peptides significantly inhibited LPS-mediated elevation of plasma IL-6 and ROS. Plasma levels of total cholesterol and triglycerides somewhat decreased in mice at 3 h post LPS, but reached statistical significance only in Ac-hE18A-NH2 group (P < 0.05 versus saline, not shown). By 27 h post-LPS, both peptide-treated groups had reduced SAA levels compared with saline-treated group (Figure 8D); however, only Ac-hE18A-NH2 reduced plasma ROS levels (Figure 8E). There was no difference in plasma cholesterol and triglycerides levels in any groups (not shown), which confirms the previous observation that the peptide Ac-hE18A-NH2 has little effect on plasma cholesterol in normolipedimic animals.
48
Ac-hE18A-NH2 reduces endotoxin activity and inflammatory response in endotoxemic mice. Either peptides (100 µg, i.v.) or saline (100 µl) were administered 5 min after LPS injection (50 µg, i.p.) in C57BL/6 mice. (A–E) LPS from E. coli O26:B6 was used for mice stimulation. Endotoxin activity (A), IL-6 levels (B) and relative change of ROS (C) were measured in plasma 3 h post-LPS. SAA (D) and ROS (E) were measured in plasma at 27 h post-LPS. Mice were fasted 3 h before blood collection. (F–H) LPS from E. coli O55:B5 was used for mice stimulation. Plasma endotoxin activity was measured at 3 h (F) post-LPS. Plasma levels of IFN-γ were measured at 3 h (G) and at 24 h (H) post-LPS. *P < 0.05, **P < 0.01 versus LPS, †P < 0.05 vs. controls, ‡P < 0.05 versus LPS plus L-4F [n = 5–6/group in (A) and (D), n = 6/group in (F) and (G), n = 7/group in (B), (C) and (E)].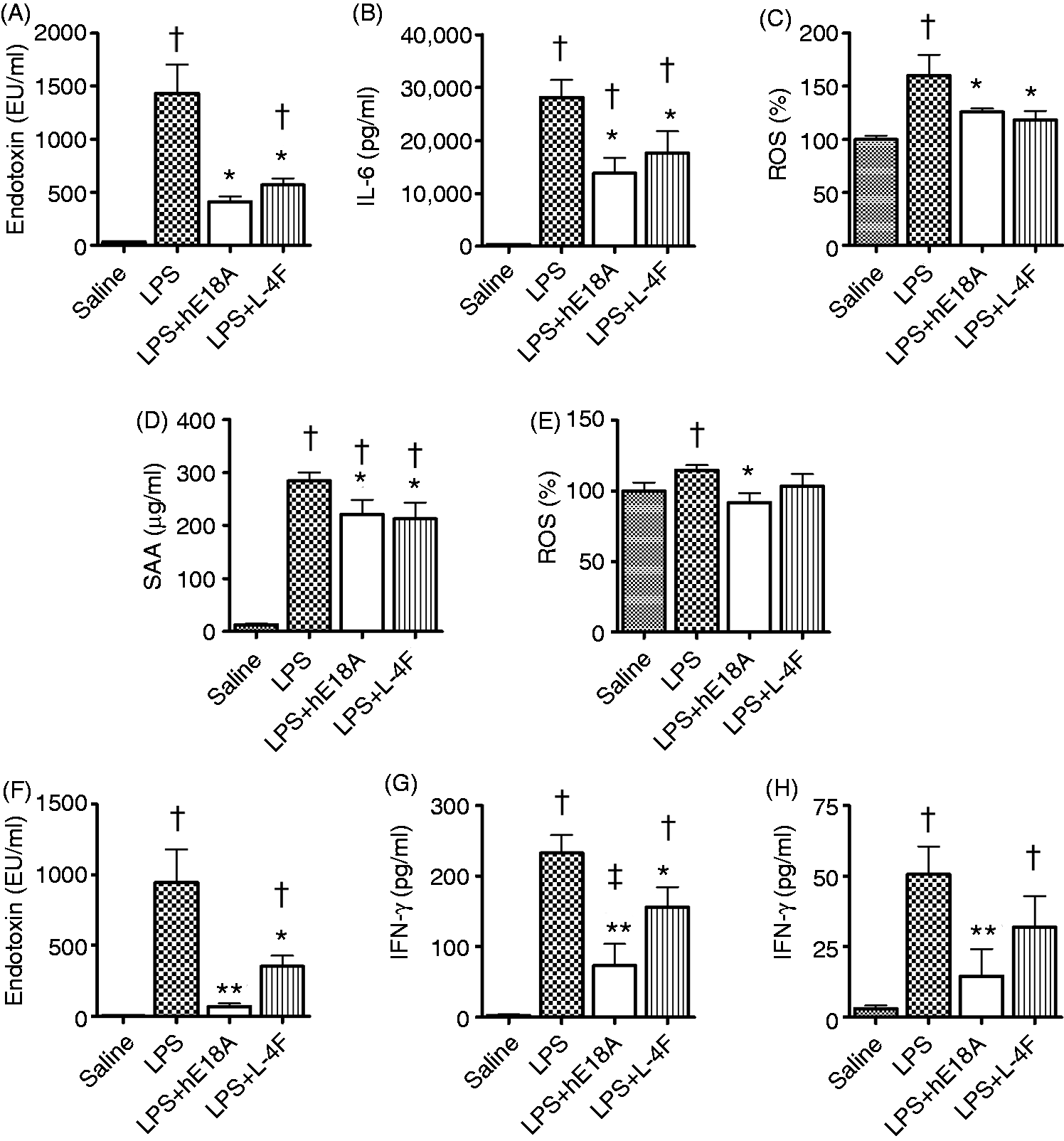
In a similar experiment, we used LPS from a different source (E. coli O55:B5) and measured changes in plasma endotoxin LAL activity and plasma levels of IFN-γ. By 3 h post-LPS, plasma endotoxin activity was highly elevated (P < 0.05 versus control mice; Figure 8F). Ac-hE18A-NH2 inhibited plasma endotoxin activity by 93% (P < 0.001 versus LPS). The effect of Ac-hE18A-NH2 on endotoxin was notably larger than the corresponding effect of L-4F, which reduced plasma endotoxin activity only by 62% (P < 0.01 versus LPS; Figure 8F). Plasma IFN-γ was significantly elevated at 3 h and 24 h post-LPS, and Ac-hE18A-NH2 prevented this elevation at both time-points, whereas L-4F reduced IFN-γ only at 3 h post-LPS (Figure 8G, H). Moreover, even at 3 h, Ac-hE18A-NH2 was more effective in reducing plasma IFN-γ than the peptide L-4F (P < 0.05 versus L-4F).
In previous animal experiments, the efficiency of L-4F, administered after LPS, to reduce LPS-induced inflammatory responses, including plasma ROS elevation, has been demonstrated.44,55 Here, we also tested the efficacy of Ac-hE18A-NH2 in neutralizing plasma endotoxin and reducing plasma ROS elevation induced by LPS (E. coli O26:B6) at different intervals. In the first setting, mice were pretreated with Ac-hE18A-NH2 (100 µg, i.v.) 30 min before LPS (25 µg, i.p.). At 3 h post-LPS, mice pretreated with saline had highly elevated endotoxin activity (P < 0.05 versus control), whereas mice pretreated with Ac-hE18A-NH2 had low plasma endotoxin activity similar to that of control mice (P < 0.05 versus LPS, not significant versus control; Figure 9A). Ac-hE18A-NH2 inhibited LPS-induced elevation of plasma ROS levels (P < 0.05 versus LPS; Figure 9B). Both LPS groups had lower cholesterol levels than control mice (P < 0.05 versus control; Figure 9C). In another experiment, Ac-hE18A-NH2 (100 µg, i.v.) was administered 3 h post-LPS injection (25 µg, i.p.). Thirty min after Ac-hE18A-NH2 administration, plasma endotoxin activity was significantly reduced (P < 0.05 versus LPS alone; Figure 9D), with no effect on plasma ROS (not shown). Additional administration of LPS and Ac-hE18A-NH2 (3 h post-LPS) to mice (every second day for a total of three times) showed significantly reduced ROS levels in the Ac-hE18A-NH2 group (P < 0.05 versus LPS; Figure 9E) 30 min post-final Ac-hE18A-NH2.
Impact of Ac-hE18A-NH2 timing on effects in endotoxemic mice. (A–C) Ac-hE18A-NH2 (100 µg, i.v.) was administrated 30 min before LPS (25 µg, i.p.). Plasma endotoxin activity (A), relative plasma levels of ROS (B) and plasma cholesterol levels (C) are measured 3 h post-LPS. (D–F) Ac-hE18A-NH2 was administered 3 h post-LPS. (D) Plasma endotoxin activity measured 30 min after single hE18A administration. (E) Relative changes of ROS in plasma of mice 30 min after the third administration of LPS and Ac-hE18A-NH2 (administered every second day, three times). (F) Plasma levels of total cholesterol 30 min after the third administration of LPS and peptide. *P < 0.05, **P < 0.01 versus LPS, †P < 0.05 and ††P < 0.01 versus controls [n = 5/group in (A), (B) and (C), n = 9/group in (D) and n = 4/group in (E) and (F)].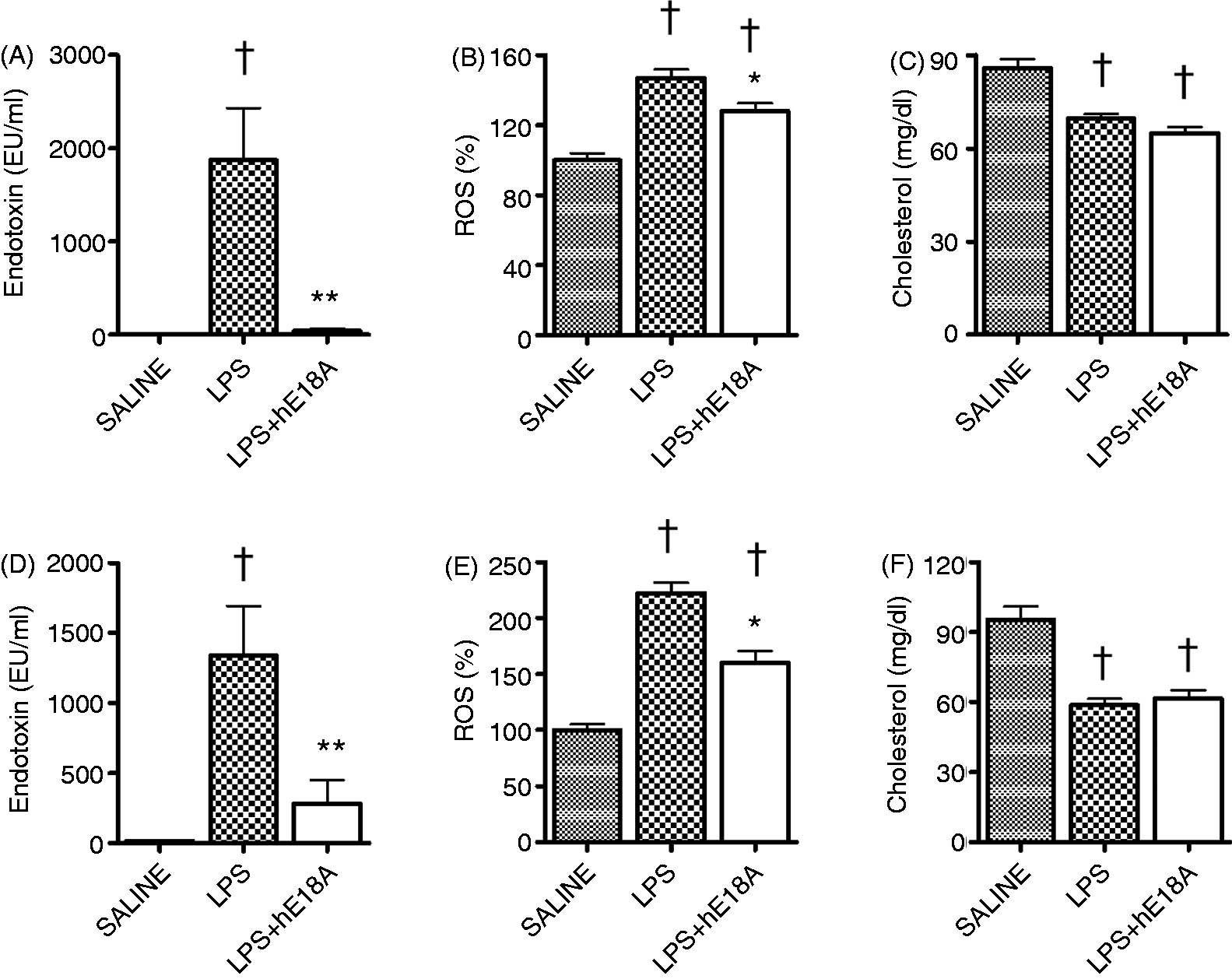
Discussion
In the present study, we demonstrate that peptide Ac-hE18A-NH2 has a direct anti-endotoxin activity and appears to exhibit stronger post LPS anti-inflammatory action than 4F. We conclude that the anti-endotoxin effect is an additional beneficial property of the plasma cholesterol-lowering apoE mimetic peptide.27,32,33,39,47–49 Rapidly increasing numbers of experimental and clinical studies suggest an involvement of LPS in the development of atherosclerosis.5–11 Humans are persistently exposed to LPS during their entire life span. In humans 61 and in animals, 62 endotoxin levels in blood increase significantly with the increased consumption of food. Chronic endotoxemia, and therefore systemic inflammation, especially owing to the Western diet, is an important pathogenic factor in the development of metabolic syndrome and atherosclerosis.16,17,61,62
In rodents, apoE has been shown to selectively bind LPS and redirect LPS from Kupffer cells to liver parenchymal cells, 51 and protects against LPS-induced lethality. 52 It has been shown that apoE-null mice are highly susceptible to endotoxemia, likely owing to a decreased LPS neutralizing capacity of apoE-deficient plasma. 63 Reconstitution of hepatic apoE expression in the liver of apoE-deficient mice normalizes the inflammatory response to low doses of LPS. 53 The apoE-deficient mouse and/or the Western diet are extensively used to study atherosclerosis. We therefore suggest that a high fat diet-induced endotoxemia,61,62 especially in the absence of endogenous apoE, 63 may contribute to systemic inflammation. This might be an important factor contributing to atherosclerosis along with hyperlipidemia in apoE-deficient mice. Therefore, anti-atherogenic effects of the apoA-I mimetic peptide 4F (which has no effect on cholesterol levels) in apoE-null mice and in LDL-receptor null mice on the Western diet 41 could, in part, be due to its strong anti-endotoxin activity. Recent studies in female apoE-null mice with already existing lesions have found that Ac-hE18A-NH2 is more effective in inhibiting lesions than 4F. 33 As the present work suggests that the anti-endotoxin activity of Ac-hE18A-NH2 is superior to 4F, such activity may also contribute to the superior effect of Ac-hE18A-NH2 in apoE-null mice. 33 However, the relative role of endogenous endotoxemia in atherosclerosis development in high-fat diet models needs to be better evaluated in future research.
The superior anti-inflammatory and anti-endotoxin activity of Ac-hE18A-NH2 to 4F in the present work can include several mechanisms. One possible mechanism is related to the cationic nature of Ac-hE18A-NH2. The α-helical amphipathic 18A peptide is zwitterionic, similar to 4F, which readily binds lipid A of LPS and neutralizes endotoxin activity.38,42,43 Addition of the hE domain to 18A significantly increases the helicity of this dual-domain molecule and increases its positively charged group. 47 As an amphipathic peptide, Ac-hE18A-NH2 is expected to readily bind to the lipid A component of LPS. In addition, the hydrophobic residues in the hE domain of Ac-hE18A-NH2 appear to extend the hydrophobic face of the 18A part of the molecule (Figure 1), which might enhance its lipid-binding property.39,49 The positive charges at the N-terminus of Ac-hE18A-NH2 also should bind to the negatively charged headgroup of LPS, analogous to polymyxin B. 49 This could result in better binding of Ac-hE18A-NH2 to wild smooth LPS like E. coli 055:B5 and better inhibition of LPS pathogen activity. 64 In our experiments, Ac-hE18A-NH2 inhibited endotoxin activity of E. coli 055:B5 more strongly than 4F peptide in vitro and in mice (although the effect was not statistically significant). Ac-hE18A-NH2 seems to also be more efficient than 4F in disrupting E. coli 055:B5 aggregates. These observations might indirectly confirm this hypothesis. The direct interaction of Ac-hE18A-NH2 with LPS is also evident from changes of electrophoretic mobility of the peptide in the presence of LPS.
However, this mechanism cannot explain all differences in Ac-hE18A-NH2 and 4F effects measured in our study, especially for E. coli O26:B6, to which both peptides seem to have similar neutralizing activity. These differences might be related to the fact that both peptides produce direct effects on cells independently of their endotoxin-neutralizing properties. L-4F has recently been shown to stimulate cholesterol efflux from human monocyte-derived macrophages, leading to cholesterol depletion and disruption of lipid rafts. 60 This resulted in down-regulation of expression of cell-surface TLRs, CD14 and CD11b, and other pro-inflammatory receptors in these cells.60,65 Dose-dependent increases in cholesterol efflux from THP-1-derived macrophages, starting with a dose as low as 5 µg/ml, have been shown for Ac-hE18A-NH2, 49 which should likely also result in lipid raft disruptions and cell modification. In addition, Ac-hE18A-NH2 can also induce secretion of cell-surface apoE and preβ-HDL formation. 49 This effect is augmented by the recycling property of Ac-hE18A-NH2 in cells. 49 Though we did not analyze the mechanisms of possible direct anti-inflammatory activity on cells, results of our experiments with peptide pretreatment in THP-1 cells suggest that such activity of Ac-hE18A-NH2 is more potent than that of 4F peptide.
In our in vitro experiments, we studied the role of serum in the mechanisms of peptide anti-endotoxin activity. Efficacy of peptides to inhibit IL-6 production by THP-1 cells did not differ in serum and serum-free media, which suggest little effect of serum on peptide effects measured in the present work. However, the effects of peptides on TNF-α levels appeared to be stronger in serum-free media than in serum, which might be accounted for by several possibilities. First, some data suggest that the production of TNF-α by THP-1 cells upon prolonged stimulation with high LPS concentration might not be affected by serum components. 51 At the same time, peptides might produce stronger effects on cell membrane and cholesterol efflux without serum lipids in cell media. In addition, peptides might bind and neutralize LPS better in serum-free media than in serum owing to lack of competition with different serum components for LPS or peptide binding.
Additional anti-inflammatory mechanisms of Ac-hE18A-NH2 could be related to its capability to bind heparan sulfate proteoglycans (HSPG). Ac-hE18A-NH2 binds HSPG on hepatocytes. 47 The maximum amount of the peptide is found in the liver. 48 It would be reasonable to suggest that Ac-hE18A-NH2 binds LPS and transports to liver (analogous to transporting LDL) and thus mimics the property of native apoE. 51 HSPG is also actively expressed on leukocyte membranes and human monocytes upon LPS stimulation. 66 HSPG is involved in the initial adhesion of leukocytes to the inflamed endothelium, the subsequent chemokine-mediated transmigration through the vessel wall, and the establishment of both acute and chronic inflammatory reactions. 67 Heparan sulfate fragments can activate TLR4-dependent signaling in cells, thus inducing the innate response. 68 Though we did not study this, Ac-hE18A-NH2 could, potentially, bind HSPG on the leukocyte membrane or HSPG fragments in cell media, and inhibit progression of the inflammatory response induced by LPS. This effect is augmented by the recycling property of Ac-hE18A-NH2 in cells. 49 Finally, the difference in clearance kinetics of Ac-hE18A-NH2 and 4F could play some role in our in vivo results. In mice, the Ac-hE18A-NH2 clearance curve is fitted to a bi-exponential equation with half-times for the rapid phase of 0.03 h and 1.61 h for the slow phase. 48 Plasma clearance of 4F in mice is also fast. 25 As measured for 3F-2 peptide with binding and bioactivity characteristics very similar to 4F, its clearance curve is fitted to an exponential equation with a half-time of only 0.54 h. 69 These additional properties of Ac-hE18A-NH2 (and not 4F) may also be involved in its enhanced ability to inhibit LPS-induced inflammation.
In the present work we did not compare the effects of peptides on cell signaling pathways activated by LPS stimulation owing to their potential complexity, which would overwhelm the scope of this work. At least two additional factors should be considered: (1) different LPS, upon TLR4 stimulation, can differently utilize MyD88-dependent and MyD88-independent signaling pathways; 70 (2) the stimulation pathways of short O-chain E. coli 026:B6 serotype can be mediated through TLR4-independent mechanisms, which might significantly differ from that of the more typical wild smooth E. coli 055:B5 serotype. 71 The effects of peptides on these possible pathways have to be a subject of further research.
In conclusion, we demonstrate here that the apoE mimetic peptide Ac-hE18A-NH2 has anti-endotoxin activity in endotoxemic mice, human whole blood, isolated human primary leukocytes, and human monocyte THP-1 cells. Ac-hE18A-NH2 appears to produce stronger post-LPS anti-inflammatory effects than the apoA-I mimetic peptide 4F, which opens prospective clinical applications for Ac-hE18A-NH2.
Footnotes
Funding
This work was supported by NIH grants NHLBI K08HL085282 (H.G.) and NHLBI HL 34343 and R01090803 (G.M.A.)
Conflict of interest
G. M. Anantharamaiah is a principal in Bruin Pharma, a startup biotechnology company. G. A. Anantharamaiah and M. N. Palgunachari hold stocks in Lipimetix Development LLC. The others authors have no potential conflicts of interest to declare.