
Abstract
Effective prevention of new HIV infections will require an understanding of the mechanisms involved in HIV acquisition. HIV transmission across the female genital tract is the major mode of new HIV infections in sub-Saharan Africa and involves complex processes, including cell activation, inflammation and recruitment of HIV target cells. Activated CD4+ T-cells, dendritic cells (DC) and macrophages have been described as targets for HIV at the genital mucosa. Activation of these cells may occur in the presence of sexually-transmitted infections, disturbances of commensal flora and other inflammatory processes. In this review, we discuss causes and consequences of inflammation in the female genital tract, with a focus on DC. We describe the central role these cells may play in facilitating or preventing HIV transmission across the genital mucosa, and in the initial recognition of HIV and other pathogens, allowing activation of an adaptive immune response to infection. We discuss studies that investigate interventions to limit DC activation, inflammation and HIV transmission. This knowledge is essential in the development of novel strategies for effective HIV control, including microbicides and pre-exposure prophylaxis.
Introduction
The Joint United Nations Programme for HIV/AIDS report for 2012—Together we will end AIDS— estimated that 34 million people worldwide were living with HIV in 2011, and 23.5 million in sub-Saharan Africa. In the same year, 2.5 million people acquired new infections, 1 highlighting the urgent need for interventions to prevent transmissions.
Most HIV transmissions occur via the mucosal surfaces of the genital and gastrointestinal tracts. 2 Women are at higher risk of HIV acquisition than men. In heterosexual discordant relationships, male-to-female HIV transmission is approximately eightfold greater than female-to-male transmission during vaginal intercourse.3–6 Several behavioural and biological factors have been proposed to explain differences in HIV acquisition between men and women, including differences in sex hormones, stage of HIV infection, age of index patient, condom use, male circumcision, presence of genital ulcers, mucosal area exposed during sexual intercourse, areas of vulnerability at the transformation zone of the cervix, dose of HIV transmitted in female versus male genital secretions, and geographical location. 4 It is estimated that 30–40% of annual worldwide infections occur through HIV invasion of the female genital tract via exposure to virus-containing semen. 7 In the female genital tract, HIV penetration and infection occurs through vaginal, ectocervical and endocervical mucosa, but the relative contribution of each site to successful transmission remains unknown. While the single-layer epithelial lining of the endocervix offers the best opportunity for HIV to cross the barrier, the relatively large surface area of the ectocervix and vagina may offer HIV a greater chance of penetrating the epithelium.7,8
The mechanisms by which HIV crosses the genital epithelium are complex. It has been suggested that during sexual transmission, cell-free or cell-associated HIV traverses the female genital mucosa through transcytosis and/or transepithelial emigration of infected dendritic cells (DC), among other routes.9,10 Transcytosis of cell-free HIV-1 across epithelial surfaces of the genital tract occurs through an epithelial transcellular vesicular pathway, where HIV-1 virions are captured on the surface of the epithelium by epithelial cells into vesicles and are liberated on the stromal side of the epithelium. 11 Many viral and host components have been implicated in this process. These include viral envelope glycoprotein (gp) 41 and gp120, glycosphingolipids, the co-receptor CCR5, and the heparin sulfate proteoglycan attachment receptors, among others. The transcytosed virus can then bind to underlying CD4+ T cells leading to an effective infection. Only a small percentage of viruses cross the epithelial barrier by transcytosis.12,13
Transepithelial migration of HIV occurs through cell-associated viruses (i.e. HIV-infected host cells). 9 Seminal cells containing HIV particles may become trapped in the mucus covering the epithelial surface. 14 Upon contact with the epithelium, these cells can release free viruses (which are able to penetrate between epithelial cells), be captured by tissue-resident DC, including Langerhans cells (LCs) and, ultimately, become internalised in the endocytic compartment.14,15 These cells may become activated and migrate into the basal compartments where they can transfer the virus to CD4+ T-cells by trans-infection. 16 During coitus, mechanical abrasions of mucosal surfaces induced by intercourse could allow free or cell-associated virus in semen to penetrate the epithelium and have direct access to DC, macrophages or CD4+ T cells that lie underneath the stroma. 7
Of the potential target cells present in the genital tract, DC, macrophages and CD4+ T-cells are the most likely cells infected with HIV infection. 17 Although all these cell types can become infected with HIV to varying degrees, CD4+ T-cells are the most susceptible to HIV infection. DC become infected with HIV both in vivo and in vitro, albeit at low levels, and can migrate to draining lymph nodes where Ag presentation occurs, leading to activation of CD8+ and CD4+ T-cells, which can, in some instances, control viral replication during established infection. 18 DC, macrophages and epithelial cells of the genital mucosa can recognise pathogens and are able to produce inflammatory cytokines or chemokines, which contribute to an immune response. The recognition of pathogens and the production of cytokines and chemokines can lead to activation and recruitment of target cells to the genital mucosa, which, although an integral process in normal host immunity, in the case of HIV, can, in turn, increase the odds of HIV capture and transmission due to HIV targeting activated cells.
In this review, we discuss causes and consequences of inflammation, and their role in HIV acquisition in the female genital tract, with a specific focus on the role of DC in driving genital tract inflammation. While this review focuses only on the role of DC in driving inflammation, it is important to note that other innate cells such as macrophages and neutrophils may also play a critical role in inflammation and HIV transmission in the female genital tract. We will discuss possible ways of regulating genital tract inflammation through this focus on DC, which may have an effect on HIV transmission.
DC subsets and pathogen binding receptors
DC were discovered in the 1970s, and today represent a diverse and rare population of leukocytes with several subsets that differ in their origin, maturation state and anatomic location.19,20 DC are derived from haematopoietic bone marrow progenitor cells and are usually found in an immature state, and are the first to respond to invading pathogens. DC become activated upon pathogen recognition leading to up-regulation of activation markers and cytokine production. 21 DC can be infected by a wide variety of pathogens, including bacteria and viruses. Recognition of pathogens by DC and subsequent infection occur through various receptors expressed on these cells. The type of innate response, mostly mediated by DC, may dictate the type of adaptive immune response to an infection with a pathogen. Activation of an adaptive response by DC is achieved through Ag presentation of pathogen-derived peptides to cells of the adaptive immune system. The ability of DC to produce cytokines and to up-regulate expression of MHC class I and II molecules and co-stimulatory markers, makes them the primary and most efficient activators of adaptive immunity. 22
DC subsets differentially express pathogen-binding receptors depending on their function and location. 23 The type of PRR triggered may determine the outcome of an innate immune response, as binding to different receptors may result in distinct immune outcomes. 24 PRRs are expressed mainly on innate cells and are centrally involved in recognition of invading pathogens and induction of immune responses.25,26 They bind to conserved structures on microbial species known as PAMPs or damage associated molecular patterns (DAMPs), which are danger signals resulting from necrotic, stressed or apoptotic cells. 27 These receptors include TLRs, NOD-like receptors (NLRs); C-type lectin receptors (CLRs), such as DC-specific intracellular adhesion molecule 3 (ICAM-3)-grabbing non-integrin (DC-SIGN); mincle, dectin-1, Man receptor, retinoic-acid-inducible protein RIG-1-like receptor, complement receptors and scavenger receptors.28,29 Of these PRRs, TLRs have been studied most extensively. There are 10 characterised TLRs (1–10) in humans, which are located either on the plasma membrane or within endosomes recognising a wide variety of PAMPs. TLRs 1, 2, 4, 5, 6 and 10 are mainly expressed on the plasma membrane and recognise microbial membrane components such as lipids, lipoproteins and proteins. TLRs 3, 7, 8 and 9 are expressed in endosomal compartments where they recognise nucleic acid components of (typically) viral origin.30,31 Twenty-three human NLR genes have been identified, but the physiological function of most of these receptors remains poorly understood.32,33 The well-characterised members of the NLR family are NOD1 and NOD2, which recognise distinct structural motifs derived from peptidoglycans. 34 Other receptors expressed by DC that are relevant to pathogen binding and signalling include chemokine receptors. 35 Chemokine receptors bind chemokines, which are necessary for recruitment of cells during inflammation and homeostasis. These receptors are expressed by leukocytes, including DC, macrophages and CD4+ T cells. Several chemokine receptors have been described with varying functions in the immune response to pathogens. 35 Here, we focus mainly on the chemokine receptors CCR5 and CXCR4 because they are co-receptors for HIV entry into target cells. 36
DC subtypes, identification markers and major HIV-related receptors expressed.
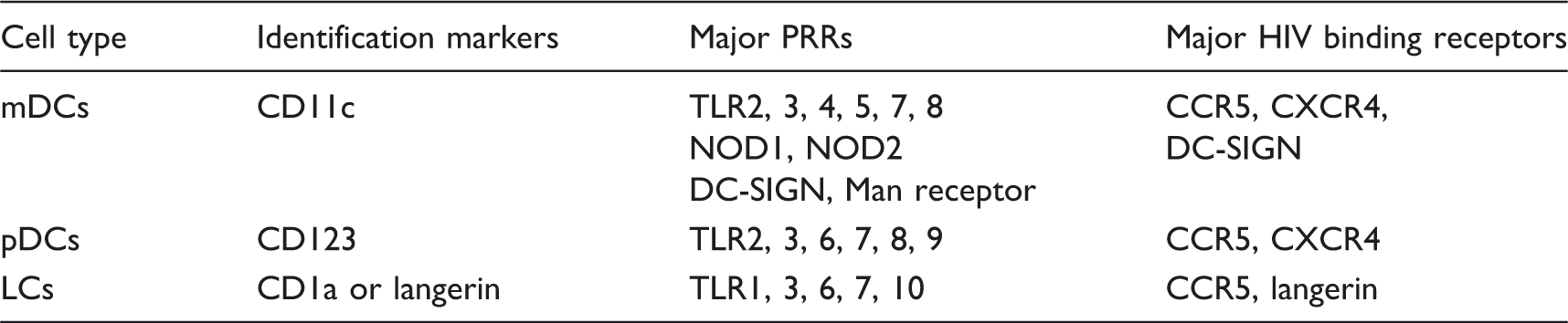
mDCs
These cells are also referred to as conventional or classical DC, and are characterised by the surface expression of CD11c (CD11chigh) and the absence of CD123 expression (CD123low) (Table 1). 37 mDC originate from common myeloid progenitors and are found in blood in an immature state. These cells migrate into tissues in response to chemotactic cytokines and contribute to the local inflammation. 38 mDC express a wide range of PRRs for the recognition and phagocytosis of pathogens, leading to the production of cytokines and chemokines, and the up-regulation of surface co-stimulatory molecules. Commonly studied PRRs include TLRs 2, 4, 5, 6, 7, 8, and NOD1 and NOD2. DC-SIGN, CCR5 and CXCR4 are also highly expressed on these cells and are of relevance to HIV infection. 39 mDC can be further subdivided into mDC1 [expressing CD11c and CD1c (BDCA-1)] and mDC2 [expressing CD11c and CD141 (BDCA-3)]. 40 These subsets are functionally distinct: CD11c + CD141 + mDC2 are more efficient than mDC1 at capturing exogenous Ags for cross presentation on MHC class I molecules to CD8+ T-cells. mDC1 and mDC2 have been reported to induce Th type 1 and Th2 T-cell differentiation respectively.41–43 mDC1 and mDC2 both express a wide range of TLRs, albeit at different levels. mDC1 express high levels of TLR4, 5 and 7, while mDC2 express high levels of TLR 3 and 8. These differences in PRR expression may explain their different roles in the immune response to infection.
pDCs
In contrast to mDC, pDC express high levels of CD123 (CD123high) and low levels of CD11c (CD11clow) (Table 1). They can also be identified by the expression of CD303 or BDCA-2.37,40 pDC originate from a common lymphoid progenitor as pre-pDC expressing CD123 and require granulocyte–macrophage colony stimulation factor (GM-CSF) to differentiate into immature cells. 44 pDC are principally found in blood and migrate to tissues in response to chemotactic cytokines and contribute to the local inflammation process. 38 pDC play a major role in the immune response to viral infections, as the major producers of anti-viral type I IFNs. pDC exhibit several additional characteristics that make them effective during viral infections.20,45 They contain MHC class I molecules in their early endosomal compartments, which facilitate Ag presentation from the exogenous processing pathway to CD8+ T-cells. In addition, the late endosomal compartment of pDC contain MHC class II molecules for viral Ag presentation to CD4+ T-cells. An extensive endoplasmic reticulum compartment facilitates high-capacity secretion of anti-viral factors, including type I IFNs. The secretion of type I IFNs, IL-6, and the expression of CD70 upon stimulation with CpG (a TLR9 ligand) make pDC efficient activators of plasma B cells and Ab responses.46,47 These cells are central to anti-viral immunity owing to the high density expression of receptors that recognise or bind viral components and production of type 1 IFNs. These receptors include chemokine receptors CCR5 and CXCR4, TLRs 3, 7 and 9.39,48 They also express other TLRs, including TLRs 1, 2, 4, 6 and 10, which can recognise bacterial components.49,50
LCs
LCs are DC found in the skin and the mucosal epithelium of the vagina and ectocervix. LCs are often defined by the expression of CD1a and/or langerin, a C-type lectin receptor (Table 1). 51 LCs are thought to originate from bone marrow haematopoietic precursors, but are maintained by local precursors present in the skin.52,53 LCs are usually the first APC to encounter pathogens in the genital tract. Their high motility allows them to emigrate from tissues to draining lymph nodes upon contact with the Ags.54,55 LCs express PRRs, including TLRs and chemokine receptor CCR5. TLRs expressed by LCs include TLRs 1, 3, 6, 7 and 10, but little or no TLR 2, 4 or 5.56,57 LCs are also characterised by the presence of Birbeck granules that are involved in the cytolytic breakdown of pathogens. 58 Birbeck granules are rod-shaped subdomain cytosolic organelles of the endocytic recycling compartment in LCs. 59
Innate receptor signalling and generation of an immune response
DC play an important role in Ag capture and presentation to T-cells, critical to immune surveillance. Upon recognition of the ligand expressed on pathogens by PRR on DC, a signalling cascade is initiated, which involves either phosphorylation or de-phosphorylation of different adaptor molecules and transcription factors. 60 This signalling cascade is necessary for the generation of an immune response, including cytokine/chemokine production and maturation of innate cells. Adaptor molecules associated with TLR signalling include MyD88, TIR-domain containing adapter–inducing IFN-β translocating chain associated membrane protein (TRIF), and TIR domain-containing adapter proteins (TIRAP).61,62 All TLRs (except TLR3) signal through MyD88. TLR3 signals through TRIF, while TLR4 can signal through either MyD88 or TRIF. All of this signalling via these adaptor molecules culminates in the activation of the transcription factor NF-κB, which translocates into the nucleus for the induction of pro-inflammatory genes. 60 The resulting inflammation is a critical early step in controlling the invading microbe while an adaptive immune response is initiated. Although TLRs are essential for protective immunity against infection, inappropriate TLR responses may contribute to acute and chronic inflammation, as well as to systemic autoimmune diseases. 25
In contrast to TLRs, NLRs and CLRs utilise different signalling pathways, typically via the caspase recruitment domain family, member 9 (CARD9). 63 NLRs can also signal via interacting protein 2 and TANK-binding kinase 1. CLRs signal via Raf1, and FcR-gamma converging to CARD9 for signalling. Other transcription factors involved in PRR signalling pathways include activator protein-1 (AP-1), MAPK and IFN regulatory factors (IRF).64–66 Activated transcription factors translocate into the nucleus and initiate transcription of immune response genes, such as those for cytokines and chemokines. The initial production of cytokines, chemokines and other molecules that result from activating these signalling pathways then serve to shape and determine the type of subsequent adaptive immune response generated. 67
Cytokines
Several cytokines mediate inflammation and innate immune responses. These cytokines include IL-12, IL-10, IL-6, IL-1β, TNF-α and IFN-α,67,68 and play important roles in the development of inflammation and the polarisation of naïve T-cells into Th1, Th2, Th17 or regulatory T-cells (Tregs).69–71 The type of PRR triggered ultimately determines the cytokines that are produced by innate cells, and therefore the outcome of the innate response. For example, the triggering of TLRs 7 and 9 on pDC by viral components leads to elevated production of IFN-α, while triggering of cytosolic receptors such as NLRs may lead to the formation of an inflammasome complex resulting in the production of IL-1β through a caspase-1-dependent pathway.72,73 Together this provides an innovative way for the immune system to link the type of pathogen to an appropriate innate and eventually adaptive immune response.
Chemokines
Chemokines are chemotactic cytokines that are important in innate immune responses to pathogens, and facilitate leukocyte migration and positioning to ensure that the correct immune cells arrive at the correct locations, and are involved in additional processes such as leukocyte degranulation. 74 There are many chemokines with diverse and overlapping functions in immunity.75–77 These chemokines include MIP-1α, MCP-1, MIP-1β, CXCL12/SDF-1 and RANTES, among others. These are secreted in response to infection and their expression can be enhanced by other inflammatory cytokines such as IL-1β. The primary role of chemokines is to create a microenvironment that acts as a chemotactic gradient, attracting immune cells from the blood to the site of the infection, which is often in tissue.78,79 Inflammatory chemokines include CCL3 (MIP-1α) and CCL2 (MCP-1), which bind to CCR1 and CCR2 respectively.
DC activation and maturation
Most DC are immature in phenotype and undergo maturation only upon pathogen encounter. Maturation comprises morphological and functional changes associated with activation of innate cells by microbial stimuli such as TLR agonists. 20 Immature DC are efficient phagocytes and, once activated, these cells mature by increasing expression of maturation markers, while, at the same time, their phagocytic capacity is reduced. Some of the better-studied maturation markers on DC include CD86, CD80, CD83 and CD40. CD80 and CD86 belong to the B7 family of co-stimulatory molecules and provide co-stimulation during Ag presentation by binding to two molecules on T-cells: CD28 for activation and CTLA-4 for inhibition of T-cell responses. 80 CD83 is a maturation marker that is expressed on DC and up-regulated upon activation. 81 CD40 belongs to the TNF receptor superfamily and is expressed by innate cells such as DC and monocytes, and binds to CD40 ligand (CD154) expressed on T-cells, thereby activating the T-cells.82,83 While CD86 and CD80 are required for T-cell activation during Ag presentation, CD40 is required for long-term DC activation, cytokine production and T-cell polarisation.80,82,84
Inflammation in the genital mucosa
Genital immune responses are similar in some ways to responses in blood or other mucosal sites, but are also distinct owing to differences in cellular composition, local cytokine milieu, microbiome complexity and microbial burden. Inflammation generally results from increased cytokine and/or chemokine production by cells that have recognised the presence of pathogens, signalling infection. The main goal of inflammation is the increased recruitment of cells to the site of infection to perform effector functions that lead to pathogen clearance. Cytokines orchestrate an inflammatory response by inducing cell death of inflammatory tissues and modifying vascular endothelial permeability. 85 They also facilitate the recruitment of more immune cells to the site of infection, thus amplifying the inflammatory response. As DC are among the first immune cells to recognise and respond to infection with genital tract pathogens, they are likely to contribute substantially to genital tract inflammation.
Sexually transmitted infections (STIs) are recognised as major causes of inflammation in the genital mucosa. Neisseria gonorrhoeae, Chlamydia trachomatis and Mycoplasma genitalium have all been well described to cause inflammation, as well as clinically important sequelae, such as pelvic inflammatory disease in women. 86 Trichomonas vaginalis and, to a lesser extent, bacterial vaginosis (not an STI but a perturbance of local microbial flora) have also been implicated in causing inflammation through several mechanisms, including the disruption of the epithelial barrier. 87 Ulcerative STIs comprising herpes simplex virus (HSV), Haemophilus ducreyi (chancroid) and Treponema pallidum (syphilis) have all been characterised as pro-inflammatory sexually-acquired conditions that manifest themselves both systemically and in the genital tract.88–90 STI-causing pathogens are either extracellular or intracellular, and activate DC through binding to surface and intracellular PRRs such as TLRs, NLRs and CLRs that recognise PAMPs on these pathogens. DC and other PRR-expressing cells in the genital tract, such as macrophages, neutrophils and epithelial cells, 91 can become activated and produce inflammatory cytokines/chemokines upon direct binding to these genital pathogens. Neisseria gonorrhoeae and C. trachomatis have both been shown to stimulate DC and monocytes in vitro leading to secretion of pro-inflammatory cytokines.92,93 Ulcerative STIs, by definition, are those that lead to breaches in the epithelial barrier. Trauma caused on genital epithelium during sexual activity, as well as vaginal practices, such as douching, may also lead to inflammation.94,95
HIV acquisition in the genital mucosa
The vaginal and ectocervical compartments are comprised of multi-layered, stratified epithelial cells lacking tight junctions, while the endocervix is protected by a single, polarised layer of columnar epithelial cells separated by tight junctions. 17 HIV enters the lower female genital tract mostly through the vagina and ectocervix, which represent an extensive surface area when compared with the endocervix. 17
Inflammation causes the migration of HIV target cells, including CD4+ DC, macrophages and T-cells, as well as other immune cells such as neutrophils, CD8+ T-cells and NK cells to the epithelium as a natural process to mediate host defence. 7 In the rabbit vaginal irritation model, vaginal inflammation was shown to enhance trafficking of immune cells to the mucosa and enhance activation of these cells, both of which have also been associated with increased risk for HIV acquisition and transcription in infected cells. 96 The presence of microbes such as STI-causing pathogens in the genital mucosa has been associated with an increased risk of HIV-1 transmission in several epidemiologic studies.97,98
DC play a dual role in determining the outcome of HIV infection
DC can facilitate HIV transmission by getting infected by HIV directly, and then transferring the virus to CD4+ T-cells. Alternatively, DC could indirectly facilitate infection of other target cells by producing cytokines that enhance the risk of infection, by either recruiting more potential target cells (increasing target cell density) or activating target cells so they are easier to infect. The fate of HIV captured by DC may depend on the activation state of these cells or on binding receptors. The expression of CCR5 and langerin on LCs enables these cells to capture, become infected by or disseminate HIV. 99 In vitro experiments using skin explants have shown that LCs are mostly susceptible to R5-tropic viruses. 100 CXCR4 expression on LCs is still controversial. This may be due to differences in sites where these cells were isolated. While expression of CXCR4 on genital tract LCs has been reported by some, 101 others have shown that CXCR4 is not expressed on these cells in the skin. 102 Others were able to induce an increase in CXCR4 expression on epithelial LCs after culture with GM-CSF. 103 Activation of LCs in vitro down-regulates langerin expression, which makes these cells more susceptible to infection. 58 In a human skin explant model, TNF-α and PAM3, a ligand for TLR2/1, have been reported to increase HIV capture by LCs, possibly in a langerin-independent manner, and increase transmission. 104 HSV-2 has been shown to decrease langerin expression on LCs or competed with HIV for binding to langerin, thereby increasing the risk of HIV transmission. 105 LCs can also play a role in inhibiting HIV transmission. HIV binding by langerin rapidly internalises the virus to Birbeck granules leading to virus degradation. When LCs capture or internalise HIV, they rapidly migrate from the epithelium to the sub-mucosal layer where they can transfer the virus to CD4+ T-cells. They can also migrate to lymph nodes where they can present HIV-derived Ags to CD4+ or CD8+ T-cells generating an adaptive immune response, which leads to the destruction of HIV-1 infected cells. 106 HIV-infected CD4+ T-cells in the mucosa can also traffic to the lymph nodes where more HIV replication occurs resulting in more infected target cells. Thus, LCs can either inhibit or promote HIV transmission, depending on their activation status and expression of langerin.
Whereas LCs reside in the genital mucosa where they mediate initial contact with HIV, mDC and pDC are predominantly found in blood and can be recruited to the tissues in response to inflammatory signals. Several studies in animal models have reported that pDC are rapidly recruited to inflamed tissues, such as genital mucosa, via high endothelial venules, where they recognize HIV, secrete IFN-α or take up Ag and migrate to draining lymph nodes for Ag presentation.107,108 HIV-1 can infect pDC directly via receptors such as CCR5, CXCR4 and CD4, resulting in IFN-α production.109–112 Alternatively, HIV may bind and enter mDC by endocytosis mediated by DC-SIGN, CCR5 and CXCR4, with CD4 as co-receptor.113,114 While blood mDC and pDC have been reported to be more frequently infected with HIV compared with monocytes and resting CD4+ T-cells in vitro, 115 other studies reported that these cells are only rarely infected with HIV-1 and are not activated by HIV to produce cytokines or mature for Ag presentation.110,116 The minimal infection of mDC in these studies was attributed to high expression of a restriction factor SAMHD1, which is a deoxynucleoside triphosphate triphosphohydrolase that prevents reverse transcription of HIV RNA. 117 The lack of mDC maturation following binding to HIV may also be due to concurrent binding to DC-SIGN, which is known to inhibit TLR stimulation.118,119 However, mDC are known to transfer HIV to CD4+ T-cells in the absence of productive infection or become dysfunctional during HIV infection.39,120
Immature DC in tissues can more efficiently capture and internalise the virus when compared to mature DC.
121
Virus captured by mature DC or retained for longer periods by maturing DC can efficiently be transferred to CD4+ T-cells through trans-infection. DC bind and internalise HIV through CCR5, CXCR4, langerin or DC-SIGN (Figure 1). The binding of HIV to one or more of these receptors may result in different outcomes. Binding of HIV-1 gp120 to DC-SIGN leads to the internalisation of the virus and transfer to the early endosomal compartment.
122
In this compartment, if not degraded, the virus can be transported to the sub-epithelial mucosa or lymph nodes where T-cells may get trans-infected through interaction with DC. Transfer of virus from DC to T-cells can also occur independently of the DC-SIGN.
123
Once HIV is internalised by DC (Figure 1), PRRs such as TLR7 and TLR9, expressed intracellularly, recognise the presence of HIV and initiate an antiviral immune response including the secretion of IFN-α and other inflammatory cytokines by pDC and DC maturation.
Relationship between inflammation, DC activation and HIV infection. Inflammation and DC activation can increase the risk of HIV transmission, while HIV acquisition by DC can also lead to DC activation and inflammation. DC acquire HIV and can either become infected or transfer the virus to T-cells, which become infected. Inflammation can enhance HIV replication in infected cells. Anti-inflammatory mediators can be used to reduce inflammation and DC activation, which leads to a decrease in risk of HIV transmission.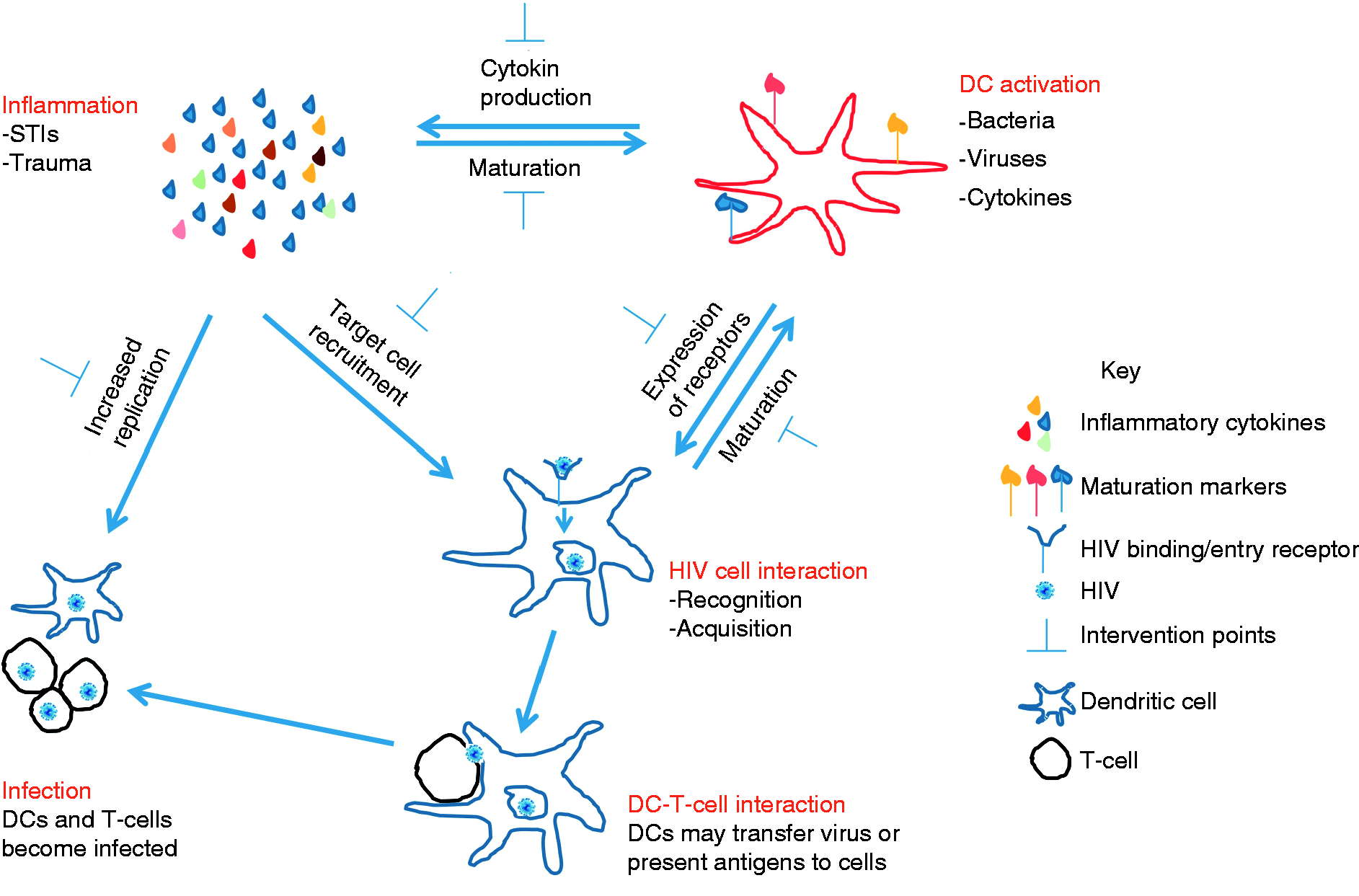
Depending on the cytokines produced by DC, and the type of cells these cytokines recruit to the genital mucosa in the presence of HIV and/or STIs, these cytokines can either be beneficial (i.e. prevent) or detrimental to (i.e. promote) HIV transmission. The production of IFN-α and IL-12 by pDC and/or mDC may result in the activation of T-cells and killing of virus-infected cells as has been shown in vitro, 124 while other cytokines such as TNF-α and IL-8 can lead to an increased replication of HIV in infected cells, and increased recruitment of target cells to the site of infection, thereby increasing the pool of susceptible target cells for newly budding HIV virions in human skin or explant cells.104,125 STIs are known to form a lethal synergy with HIV, where HIV can exploit the inflammation caused by STIs to propagate itself and establish infection. Thus, inflammation in the genital tract resulting from recognition of pathogens is a double-edged sword that needs to be tightly regulated. While genital inflammation may increase risk for HIV infection, the process of inflammation is necessary in the control of infection with other mucosal pathogens such as tuberculosis and HPV.126,127 The relative contribution of each leukocyte to the general inflammation in the genital tract is unclear and is an important area for future studies. Current evidence suggests that genital tract inflammation may be more of a ‘foe’ than a ‘friend’ in the context of HIV transmission. A fine balance between the level of inflammation that is beneficial or detrimental to host immunity against pathogens may need to be established.
The role of sex hormones in DC activation and HIV acquisition in women
Gender differences in risk for HIV infection and pathogenesis following infection may be attributed to several factors, including sex hormones. Differences in HIV shedding in genital secretions and transcription of HIV genes have been associated with the menstrual cycle in women and levels of progesterone.128,129 Hormones may have both protective and enhancing roles in HIV infection, which may be related to changes in immune cell function, as well as thickness of the epithelial lining. In macaques, administration of estriol prior to simian immunodeficiency virus (SIV) challenge was shown to thicken the vaginal epithelial resulting in protection against SIV infection. 130 Oestrogen has also been shown to directly reduce the susceptibility of CD4+ T-cells and macrophages to HIV infection in vitro through binding to oestrogen receptors and altering HIV entry into these cells. 131 Progesterone containing depot medroxyprogesterone acetate is a commonly used injectable contraceptive in high HIV prevalence regions, particularly in sub-Saharan Africa. This hormone was shown to significantly reduce the production of inflammatory cytokines by peripheral blood DC in response to TLR stimulation, as well as prevented down-regulation of CCR5 and CXCR4 on activated T-cells, and increased HIV transcription in in vitro cell cultures. 132 Women using this hormone also expressed lower cytokine levels in blood and genital secretions than non-users. 132 Hormonal contraceptive use was associated with increased risk of HIV transmission in younger women compared with older women, possibly owing to increased cervical ectopy in younger women.133,134 Hormonal contraceptive use and fluctuations in hormonal levels during the menstrual cycle lead to changes in female genital tract immunology and epithelial thickness, which may have serious consequences on risk of HIV transmission.
Reducing inflammation to prevent HIV transmission
If genital inflammation is associated with increased risk of HIV transmission, strategies to safely regulate inflammation may be able to curb the spread of HIV. These strategies could involve preventing, identifying and treating the causes of genital tract inflammation, inducing more natural anti-inflammatory mediators, or using exogenous anti-inflammatory drugs. As one of the major causes of genital tract inflammation is STIs, and most of them are treatable, active diagnosis and treatment of individuals with STIs could have a profound impact on HIV transmission. Despite this, population-wide approaches to STI prevention and treatment have demonstrated mixed efficacy in achieving this goal, and have proven to be operationally difficult to implement effectively.135–141 HSV is possibly the exception, as it infects a wide range of cells, including leukocytes, epithelial cells and nerve cells. The virus can remain latent in nerve cells until becoming reactivated when the immune system is suppressed. 142 It has been reported that even after treatment of HSV lesions and healing, HSV-induced inflammation still persists in the genital tract for up to 20 wk. 143 A vaccine to prevent HSV acquisition in adolescents could have a huge impact on preventing subsequent rates of HIV infection.
Natural anti-inflammatory mediators that balance the potential harm caused by inflammation include soluble TNF receptors (sTNF-Rs), IL-1 receptor antagonist (IL-1Ra), IL-10 and Tregs. 144 Among the anti-inflammatory regulatory molecules, the role of the IL-10 family of cytokines in human immunology and in the response to viral and bacterial pathogens has been well described. 145 IL-10 is secreted by leukocytes including macrophages, monocytes, DC and T-cells. 144 Innate cells secrete IL-10 in response to TLR stimulation and during clearance of apoptotic cells, while T-cells secrete IL-10 in response to T-cell receptor triggering.146,147 IL-10 functions by binding to the IL-10 receptors (IL-10R1 and IL-10R2), and uses the Janus kinase family members and signal transducers and activators of transcription (STAT) transcription factors to mediate its effects. 148 The main inhibitory effects of IL-10 are exerted on monocytes/macrophages by inhibiting the production of pro-inflammatory cytokines and down-regulating the expression of MHC II and CD86 in response to LPS or IFN-γ stimulation.149–151 We have also observed that IL-10 inhibits IL-6 and CD40 expression by monocytes and DC in response to TLR7/8 and TLR4 ligands. 152 IL-10 also inhibits T-cell proliferation and cytokine production by both Th1 and Th2 cells.153,154 The inhibitory effect of IL-10 on chlamydia-induced cytokine production by human epithelial cells has also been described. 155 In the context of antiviral responses, IL-10 decreases the expression of MHC I on DC, and increases deletion of mature DC by NK cells. 156 Culturing mature DC with HIV resulted in a significant increase in IL-10 production leading to a decrease in expression of CD83, HLA-DR and HLA class I, while the opposite effect was observed when immature DC were co-cultured with HIV. 156 However, IL-10 enhances the expression of other regulatory molecules such as IL-1Ra and sTNF-R.157,158
IL1Ra and sTNFR both directly inhibit the stimulatory effects of IL-1 and TNF-α via receptor competition. IL-1Ra is a member of the IL-1 family and is a naturally occurring anti-inflammatory protein that competitively blocks the binding of IL-1 to type I and II IL-1 receptors without inducing any signalling activity or intracellular response. 159 The action of TNF-α is mediated by two receptors (TNF-R1 and TNF-R2). 160 sTNF-Rs serve as antagonists of cell surface receptors for TNF-α and have a dual role in the control of inflammation in this environment. Binding of sTNF-Rs to TNF-α can either reduce the ‘toxic’ effects of TNF-α or serve as a carrier and stabiliser of TNF-α. Thus, natural immune regulatory molecules are necessary and present to control immune activation and inflammation.
The female genital tract is in constant contact with both commensal and introduced microbes, most of which do not elicit an active immune response. Because of the commensals and its primary role in fertility, this mucosal environment is immune privileged (tolerogenic), particularly with respect to T-cell populations. Tregs expressing FoxP3 and CD25 are a subset of T-cells that form part of the natural anti-inflammatory mechanisms and can actively suppress both cellular activation and inflammation via direct and indirect mechanisms. Tregs actively suppress the activation or persistence of an immune response, and prevent pathological self-reactivity that leads to autoimmune disease, by mechanisms that include IL-10 and TGF-β secretion.161–165 While this subset has not been well-characterised in the female genital tract of humans, this population is common at other mucosal surfaces, such as the gut. 166 Treg development is opposed to that of another inflammatory Th subset called Th17 cells. Both Treg and Th17 subsets are required for effective host immunity, and their relative balance is likely to influence levels of inflammation. Both of these T-cell subsets are also targets of HIV. 167 In addition to Tregs, pathogen-specific T-cells can up-regulate molecules with regulatory functions, such as IL-10 and CTLA-4, limiting their inflammatory potential. Therefore, the relevance of the Th subset balance in HIV-exposed mucosa includes their influence on the inflammatory status of the mucosal environment, and their presence as HIV targets. HIV infection is known to impair the capacity of DC to induce Tregs by limiting CD25 and FOXP3 expression in a caspase-dependent manner, possibly owing to preferential killing of Tregs by HIV-infected DC. 168 Tregs can also down-modulate the capacity of DC to activate effector T-cells through IL-10, and inhibition of the expression of co-stimulatory molecules on DCs.169–171 In combination with vaccines and microbicides to prevent infection, immune modulatory strategies that induce Tregs that dampen inflammation may have beneficial effects by limiting HIV transmission.
Natural anti-inflammatory mediators are ultimately unable to sufficiently control high genital tract inflammation in the presence of STIs or other inflammatory conditions to reduce HIV transmission. The addition of exogenous anti-inflammatory mediators is an intervention that may further augment the body's natural defences. Anti-inflammatory medication, including corticosteroids and non-steroidal anti-inflammatory drugs, have been licenced for use in humans to treat various inflammatory conditions,172,173 and may possibly have a role to play in reducing genital tract inflammation, and HIV transmission. MAbs have also been used to limit inflammation especially in autoimmune conditions, for example the use of Infliximab and other anti-TNF mAbs have clinical benefit in Crohn's disease.174–176 Although these strategies to dampen inflammation by TNF-α are useful to reduce symptoms of autoimmunity, it should be noted that important side effects have been described for anti-TNF-α treatment, which include reactivation of latent tuberculosis, pneumonia, meningitis and sepsis among others. 177
Anti-inflammatory mediators can exert the following effects: (i) a decrease in pro-inflammatory cytokine production, (ii) a decrease in the activation of DC and T cells, and (iii) a reduction in the expression of some binding/entry receptors (Figure 1). Decreases in inflammation, receptor expression and activation of DC and other target cells can have profound effects on HIV transmission in the female genital tract. 98 Animal models have shown that glycerol monolaurate (GML), a compound with anti-inflammatory properties, can inhibit SIV transmission in the genital tract. 178 GML exhibits its anti-inflammatory effect by inhibiting immune activation and cytokine/chemokine production such as IL-8 and MIP-3α, thereby reducing DC recruitment to the genital tract.178–180 Because of these anti-inflammatory properties, GML was evaluated for its ability to prevent SIV acquisition in the genital tract of rhesus macaques. All GML-exposed animals were completely protected from repeated high dose SIV challenges compared to no protection in the untreated control animals. 178 These experiments support a model in which the initial inflammatory cascade forms a central feature of subsequent risk of HIV acquisition. The use of anti-inflammatory agents in HIV control in humans should therefore be investigated further.
Conclusion and future perspectives
DC activation results in cytokine production and inflammation, and inflammation can reciprocally fuel DC activation. Both immune activation and inflammation are risk factors for HIV transmission. PRRs and chemokine receptors are centrally involved in the process of HIV recognition and acquisition. Inflammation that results from the presence of HIV and other pathogens in the genital mucosa can be regulated by various anti-inflammatory mediators—whether they are induced naturally or exogenous. Until recently, the use of microbicides to prevent HIV transmission has been largely ineffective. Some of the earlier microbicides, such as nonoxynol-9 (N-9) and cellulose sulphate, increased risk of HIV acquisition by causing genital inflammation or micro-abrasions.181,182 The new generation microbicides, which include potent anti-retroviral drugs such as 1% tenofovir topical gel showed moderate, but encouraging, success in reducing risk of HIV acquisition, although efficacy of this approach has been shown to be influenced by pre-existing genital inflammation. 183 Women in this 1% tenofovir gel trial with pre-existing genital inflammation were at increased risk of HIV acquisition, irrespective of whether they were using the tenofovir gel or placebo. 184 Strategies to prevent HIV infection at the genital mucosa (such as vaccines or microbicides) may be substantially improved on safely limiting inflammation at the genital mucosa, thereby reducing the risk of HIV transmission.
In summary, DC activation, inflammation and HIV transmission are interrelated. Whereas cell activation and inflammation may be beneficial in controlling other pathogens, these host responses inadvertently increase the risk of HIV transmission by increasing the availability of HIV target cells in the female genital tract. Identifying and treating causes of genital tract inflammation by adding anti-inflammatory agents or mediators to the female genital tract could make an important contribution to controlling HIV transmission. The counter side to this approach is that inflammation in the genital mucosa serves an important function in protection against other pathogens. More effective strategies to control HIV transmission in the female genital tract will entail a comprehensive understanding of factors—cell activation and inflammation in particular—that lead to sub-optimal efficacy of anti-HIV microbicides or vaccines.
Footnotes
Funding
MSS is supported by the Columbia University-Southern African Fogarty International Training and Research Program (AITRP) funded by the Fogarty International Center, National Institute of Health (grant # D43TW00231).
Conflict of interest
The authors do not have any potential conflicts of interest to report.