
Abstract
Sepsis-induced immune reactions are reduced in TNF receptor 2 (TNFR2)-deficient mice as previously shown. In order to elucidate the underlying mechanisms, the functional integrity of myeloid cells of TNFR2-deficient mice was analyzed and compared to wild type (WT) mice. The capacity of dendritic cells to produce IL-12 was strongly impaired in TNF-deficient mice, mirroring impaired production of IL-12 by WT dendritic cells in sepsis or after LPS or TNF pre-treatment. In addition, TNFR2-deficient mice were refractory to LPS pre-treatment and also to hyper-sensitization by inactivated Propionibacterium acnes, indicating habituation to inflammatory stimuli by the immune response when TNFR2 is lacking. Constitutive expression of TNF mRNA in kidney, liver, spleen, colon and lung tissue, and the presence of soluble TNFR2 in urine of healthy WT mice supported the conclusion that TNF is continuously present in naïve mice and controlled by soluble TNFR2. In TNFR2-deficient mice endogenous TNF levels cannot be balanced and the continuous exposure to enhanced TNF levels impairs dendritic cell function. In conclusion, TNF pre-exposure suppresses secondary inflammatory reactions of myeloid cells; therefore, continuous control of endogenous TNF by soluble TNFR2 seems to be essential for the maintenance of adequate sensitivity to inflammatory stimuli.
Introduction
TNF, a key cytokine induced by inflammatory stimuli, has a wide range of biological functions. This is probably owing to the fact that it exists in two biologically active forms, as a soluble and as a trans-membrane molecule. In addition, the two forms interact differentially with two types of receptors, TNF receptor type 1 (TNFR1) and TNF receptor type 2 (TNFR2), which activate different signaling pathways. 1 Both receptors can be detected on the surface of a number of different cell types, with TNFR1 being constitutively expressed on many cell types, while the expression of TNFR2 is highly regulated and mainly restricted to immune cells. 2 In addition, the two TNFRs can be cleaved off membranes and act as soluble TNF scavengers. TNFR2, in particular, is released in large quantities during inflammation and acts as functional inhibitor of TNF.
TNFR1 is the principal receptor on most cell types, triggering inflammatory signals classically attributed to TNF, such as triggering cytotoxic and inflammatory processes.3,4 Important TNFR1-dependent signaling pathways include pathways that result in activation of NF-kB, Jun N-terminal kinase, MAPK and others. Consequently, TNFR1-deficient mice are resistant to endotoxic shock and demonstrate an impaired defense to bacterial pathogens.5,6
Soluble TNFR2 is generally considered to attenuate TNF responses by scavenging TNF. 4 The physiological role of signal transmission through TNFR2, however, is not entirely clear and genetic ablation of the gene for TNFR2 did not result in a mouse line with a strong phenotype. Membrane TNFR2 was found to enhance TNFR1-induced apoptosis, first believed to be due to a phenomenon called ‘ligand passing’ 7 and, later, to intracellular signaling.8,9 With respect to inflammatory pathways, both synergistic and antagonizing activities of TNFR1 and TNFR2 have been described. The differential interaction of membrane-bound versus soluble TNF with TNFR1 and/or TNFR2, respectively, led to the conclusion that both TNF receptors might cooperate in TNF-induced inflammatory responses.10,11 However, the precise role of TNFR2 in sepsis, where TNFR1 definitely is responsible for the inflammatory reaction as outlined above, has not been clarified. Interestingly, TNFR2-deficient mice were resistant to changes of the immune status induced by sepsis, such as increased susceptibility to subsequent infections 12 and increased numbers of regulatory T cells. 13
We used TNFR2-deficient mice to investigate whether TNFR2 might play a decisive role modulating immune responses after cecal ligation and puncture (CLP), a mouse model for induction of poly-microbial peritonitis. Assuming that dendritic cells (DC), which play a central role in linking innate with adaptive immune responses, provide a clue for the function of TNFR2, we compared primary DC from TNFR2-deficient and wild type (WT) mice and detected impaired Ag presenting capacity of TNFR2-deficient DC after CLP. This phenotypic characterization of DC should help to mechanistically understand and interpret previous observations made in experimental mouse models using TNFR2-deficient mice.
Material and methods
Animals
C57BL/6 mice were purchased from Janvier (Le Genest, France). TNFR2-deficient mice (C57BL/6-Tnfrsf1btm1Mwm) 14 were purchased from The Jackson Laboratory (Bar Harbour, ME, USA), and deficiency of TNFR2 expression was verified by PCR. Mice were housed in the animal facility of the University of Regensburg and handled in accordance with institutional guidelines.
Cells
Peripheral mononuclear cells were isolated from blood by density gradient separation. Blood containing EDTA was placed onto Pancoll (PAN-Biotech, Aidenbach, Germany) and interphase was harvested after centrifugation. Following washing, cells were placed in culture. CD11c-positive cells were isolated by magnetic-activated cell sorting (MACS) from splenic single cell suspensions following the manufacturer’s protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). For generation of bone marrow-derived DC (BMDC), mice were killed and femura and tibiae dissected, quickly placed in 70% aqueous ethanol, both ends cut and bone marrow flushed out with PBS. Cells were washed once with medium and re-suspended in RPMI medium containing granulocyte–macrophage colony-stimulating factor (GM-CSF) for generation of BMDC as described previously. 15
CLP
Mice were anesthetized (75 mg/kg Ketanest; Parke, Davis & Company, Munich, Germany) and 16 mg/kg Rompun (Bayer AG, Leverkusen, Germany) i.p. The cecum was exteriorized and 30% of the distal end was ligated and punctured once with a 27-G needle to achieve a sublethal CLP. 16
Flow cytometry
Following lysis of red blood cells (RBC) in blood and splenic single cell suspensions, cells were incubated with Abs against CD16/32 to block non-specific Ab binding and stained with fluorochrome-labeled Abs. Intracellular cytokines were detected using the Cytofix/Cytoperm kit (BD Biosciences, Heidelberg, Germany). Fluorochrome- or biotin-labeled Abs detecting mouse CD11c, CD11b, CD4, CD8a, B220, Ly6C, Ly6G, CD86, CD80, IL-12p40, IFN-γ and MHC-II, as well as fluorochrome-labeled streptavidin, were purchased from BD Biosciences, eBioscience (Frankfurt, Germany), Miltenyi Biotec (Bergisch Gladbach, Germany) or Invitrogen (Darmstadt, Germany). Cells were measured on a BD LSR-II or a BD FACSCalibur flow cytometer, and analyzed using the FacsDiva or CellQuest softwares (BD Biosciences).
Cytokine production
Splenic single cell suspensions after lysis of RBC or BMDC were cultured with cell culture medium (RPMI 1640 with 10% FCS, Pen/Strep, 50 µM 2-ME) at 37℃. For detection of IL-12p40-producing cells, cells were cultured for 4 h in the presence of Monensin (GolgiStop; BD Biosciences), usually stimulated with 5 µg/ml cytosin–guanosin oligonucleotid ODN 1668 (CpG; Metabion, Munich, Germany), but without CpG stimulation in the LPS tolerance experiments. For IL-12 family mRNA detection, cells were stimulated with CpG for 6 h. ELISA kits were used for quantification of IL-12p70 (Bender MedSystems, Vienna, Austria) in supernatants of 18-h cultures with CpG, and for quantification of IFN-γ (R&D Systems, Wiesbaden, Germany) in serum.
Ag presentation
CD11c+ cells were isolated from splenocytes by magnetic separation with anti-CD11c beads, and CD4+ T cells were isolated from splenocytes of OT-II mice with anti-CD4 beads according to the manufacturer’s instructions (Miltenyi Biotec). OT-II CD4+ T cells were labeled with 1 µM 5,6-carboxyfluorescein succinimidyl ester (CFSE) and 2 × 105 cells were incubated with 4 × 104 CD11c+ cells and varying concentrations of ovalbumin (OVA). CFSE dilution was analyzed after 72 h of culture by flow cytometry to quantify proliferation.
Quantitative real-time PCR
RNA was prepared from samples using the Nucleospin RNA II kit according to the manufacturer’s instruction (Macherey-Nagel, Düren, Germany) and cDNA was generated by transcription with Moloney murine leukemia virus reverse transcriptase (MMLV), (Promega, Mannheim, Germany). Primers for IL-12p35 mRNA (forward: TGT CAA TCA CGC TAC CTC CTC; reverse: TTT TCT CTG GCC GTC TTC AC), IL-12p40 mRNA (forward: TCA GGG ACA TCA TCA AAC CA; reverse: CTT TCT GGT TAC ACC CCT CCT), TNF mRNA (forward: CCC CAA AGG GAT GAG AAG TT; reverse: CAC TTG GTG GTT TGC TAC GA), 18S rRNA (forward: GTA ACC CGT TGA ACC CCA TT; reverse: CCA TCC AAT CGG TAG TAG CG) and β-actin (forward: TCA CCC ACA CTG TGC CCA TCT ACG A; reverse: GGA TGC CAC AGG ATT CCA TAC CCA) were purchased from Metabion (Martinsried, Germany). Real-time PCR was performed with the iQ SYBR Green Super Mix (BioRad, Munich, Germany) on an iQ5 iCycler (BioRad) with 40 cycles (20 s at 95℃, 30 s at 58℃, 30 s at 72℃).
Mouse models
LPS tolerance model. 17
Mice were challenged 24 h after pre-treatment with either LPS from Escherichia coli 0127:B8 or human TNF (0.025 mg/mouse, i.v.) with LPS (0.1 mg/mouse, i.v.), and spleen was sampled 90 min later.
Propionibacterium acnes sensitizing model
Dead Propionibacterium acnes was injected (0.02 g/kg in 0.2 ml PBS, i.v.) and 7 d later mice were challenged with LPS from Salmonella Abortus Equi (0.01 mg/kg in 0.2 ml PBS, i.v.). Four h after LPS injection serum IFN-γ levels were determined.
Statistics
Unpaired Student’s t-test or two-way ANOVA with Bonferroni post hoc test were used in experiments with two or more experimental groups. P < 0.05 was accepted as significantly different. All statistics were performed using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA). All data shown in figures are representative of 2–3 experiments (except for Figure 2a, which was done once).
Results
Sepsis reduces lymphocyte population in TNFR2-deficient mice
Splenic lymphocyte populations in naive and septic WT and TNFR2-deficient mice.
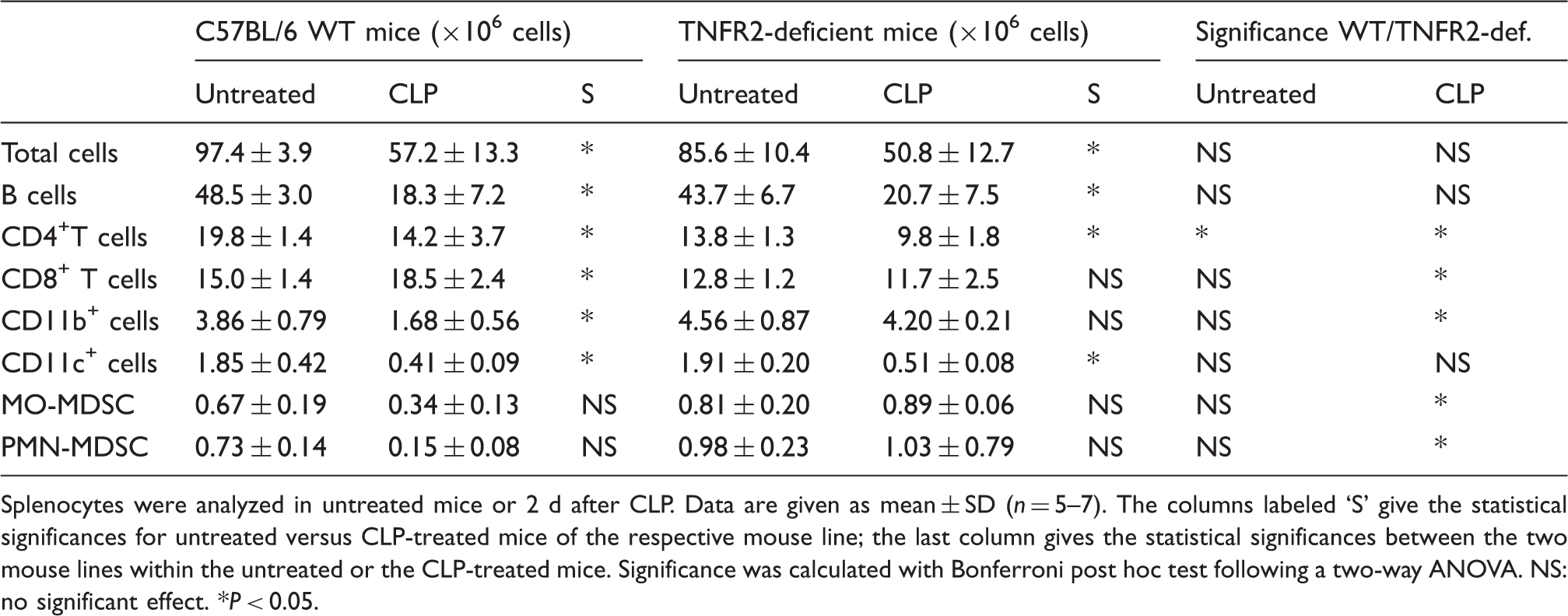
Splenocytes were analyzed in untreated mice or 2 d after CLP. Data are given as mean ± SD (n = 5–7). The columns labeled ‘S’ give the statistical significances for untreated versus CLP-treated mice of the respective mouse line; the last column gives the statistical significances between the two mouse lines within the untreated or the CLP-treated mice. Significance was calculated with Bonferroni post hoc test following a two-way ANOVA. NS: no significant effect. *P < 0.05.
Activation markers of myeloid cells in naïve and septic WT and TNFR2-deficient mice.
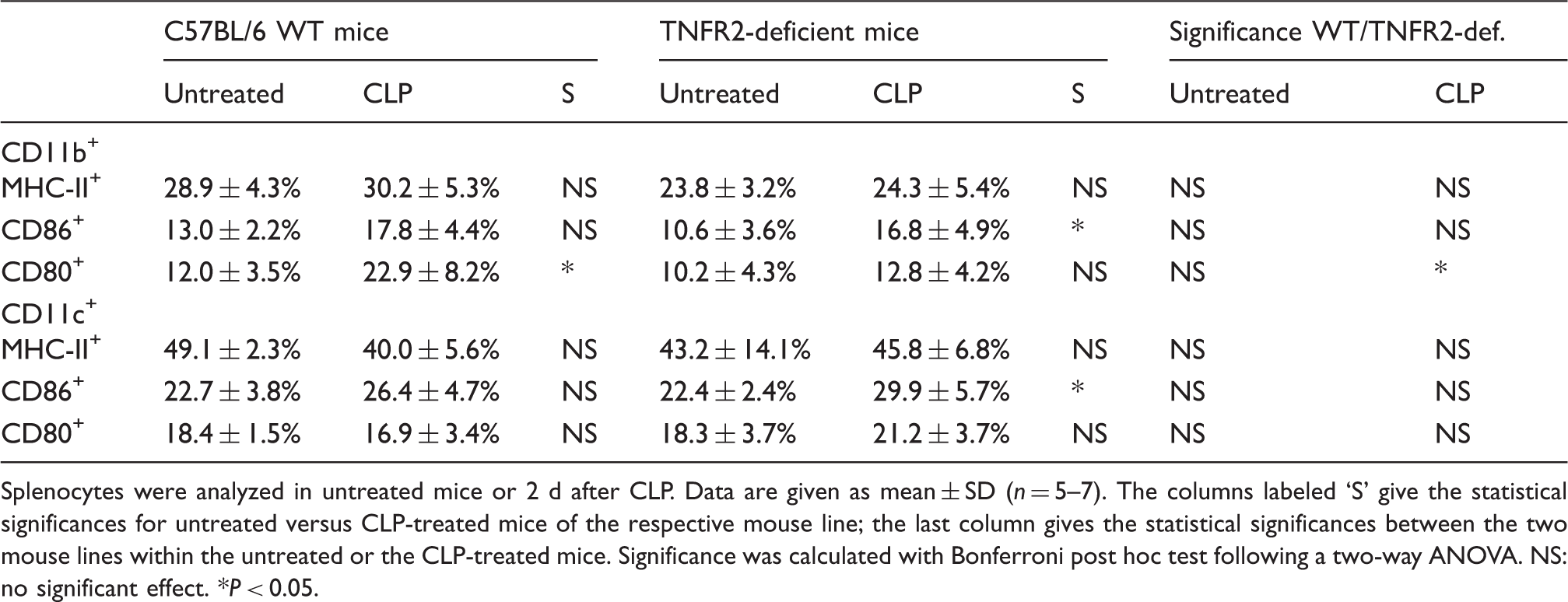
Splenocytes were analyzed in untreated mice or 2 d after CLP. Data are given as mean ± SD (n = 5–7). The columns labeled ‘S’ give the statistical significances for untreated versus CLP-treated mice of the respective mouse line; the last column gives the statistical significances between the two mouse lines within the untreated or the CLP-treated mice. Significance was calculated with Bonferroni post hoc test following a two-way ANOVA. NS: no significant effect. *P < 0.05.
Production of IL-12 of splenic DC from TNFR2-deficient mice is impaired
Next, we analyzed the impact of CLP and TNFR2 on DC function by harvesting DC 2 d after CLP and activating them in vitro. Stimulation of splenic DC with CpG resulted in lower levels of IL-12p70, as well as in reduced numbers of IL-12p40-producing DC in naïve TNFR2-deficient compared with naïve WT mice (Figure 1a, b). Also, the fraction of IL-12-producing DC was found to be similarly reduced in CpG-stimulated DC from blood of naïve TNFR2-deficient mice (Figure 1c).
WT or TNFR2-deficient mice were either left untreated or treated with CLP (naïve WT: filled circles; CLP-treated WT: open circles; naïve TNFR2-deficient: filled squares; CLP-treated TNFR2-deficient mice: open squares). MACS-purified DC were prepared from splenocytes 2 d after CLP (a, b, d) or blood cells were prepared from naïve mice (c) and cultured in the presence of CpG. (a) IL-12p70 was determined in supernatant after 18 h of culture. (b, c) IL-12p40+ DC were detected after 4 h of culture. (a–c) Each symbol represents a mouse; line indicates mean. *Statistically significant difference. (d) mRNA levels were determined after 6 h of culture. Bars indicate the mean mRNA levels of technical replicates. (e) MACS-purified splenic DC were cultured in presence of CFSE-labeled OT-II CD4+ T cells and varying concentrations of OVA. 3d, proliferation of CD4+ T cells was analyzed. Symbols indicate the mean of technical replicates.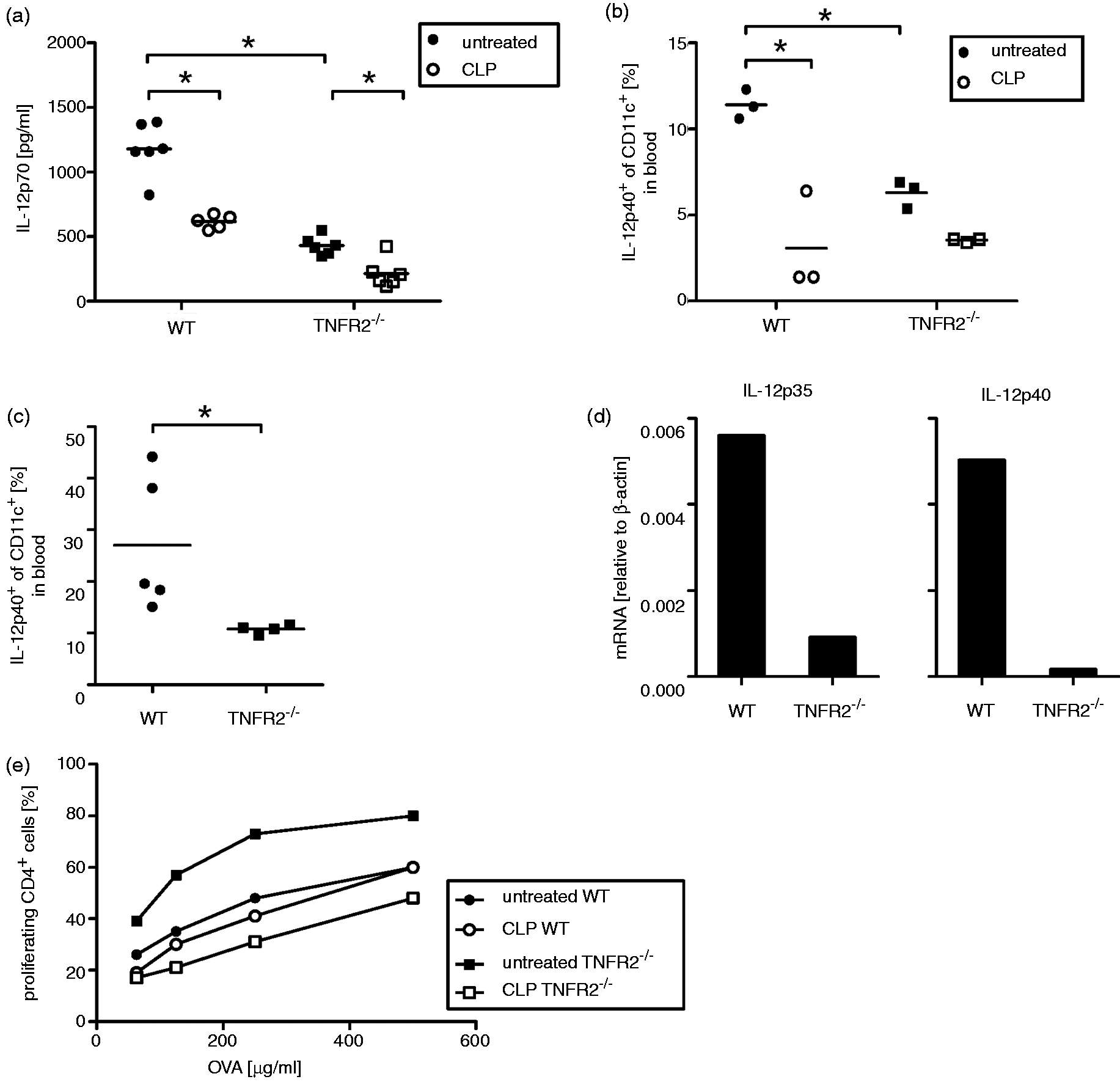
Sepsis lowered the percentage of IL-12p40-positive splenic DC and significantly reduced the production of IL-12p70 by DC isolated from spleens of WT mice, as previously described.20,21 Even though TNFR2-deficient splenic DC started off at a much lower level when derived from naïve mice, an additional sepsis-induced reduction in IL-12p40-positive splenic DC ratio and IL-12p70 production was seen in DC from septic TNFR2-deficient mice (Figure 1a, b). This CLP-induced reduction in IL-12p70 in TNFR2-deficient mice was still significant, while no significant reduction was measured in the percentage of IL-12p40-positive CD11+ cells. CpG-induced mRNA expression levels of proteins constituting the IL-12 family (p40, p35, p19, p28 and Ebi 3) were determined. All tested genes, and, in particular, genes coding for p40 and p35, were found to be more strongly expressed in DC from naïve WT than in naïve TNFR2-deficient mice, supporting the IL-12 protein data (Figure 1d and data not shown).
As it was previously shown that CLP leads to epigenetic changes in DC, 21 we wondered if the reduced IL-12 production by DC from TNFR2-deficient mice is based on similar alterations. To investigate if precursor cells of DC in the bone marrow are already destined to have an IL-12 production deficiency owing to (possibly epigenetic) alterations, BMDC from both mouse lines were generated by culturing bone marrow cells in the presence of GM-CSF. The resulting BMDC from the two mouse lines showed equal IL-12 production after CpG stimulation (data not shown) and not the reduced IL-12 production phenotype seen with splenic DC from TNFR2-deficient mice. The changes in IL-12 production could be based on the lack of TNFR2 on the DC themselves, or on a general change in the microenvironment in TNFR2-deficient mice. In order to clarify this, we generated chimeric mice with a TNFR2-deficient hematopoietic system in a WT host. Splenic DC of these chimeric mice did not show impaired IL-12 production capacity: splenic DC of WT-in-WT control chimeric mice stimulated ex vivo with CpG showed 4.6 ± 0.5% IL-12p40-producing cells, and splenic DC from TNFR2-deficient-in-WT chimeric mice demonstrated 5.1 ± 1.0% IL-12p40-producing DC after CpG stimulation. Similarly, BMDC created from these chimeric mice did not show a difference in IL-12-production (261.3 + 2.3 pg/ml in WT-in-WT and 225.6 + 147 pg/ml IL-12p70 in TNFR2-deficient-in-WT). These data indicate that the reduced IL-12 production of splenic DC in TNFR2-deficient mice is an acquired feature and not a genetically pre-determined phenotype.
Ag presentation is not diminished in TNFR2-deficient DC
Besides IL-12 production, activation of T cells is an important function of DC. We have previously shown that sepsis induced by CLP impaired the capacity for Ag (OVA) presentation of splenic DC from WT mice resulting in reduced T cell proliferation. 18 Surprisingly, Ag-presenting splenic DC from naïve TNFR2-deficient mice showed an enhanced capacity for induction of T cell proliferation compared to DC from WT mice (Figure 1e). However, CLP markedly reduced the T cell activation capacity of DC from TNFR2-deficient mice, demonstrating a pronounced sepsis-induced suppressive effect by splenic TNFR2-deficient DC. The sepsis-induced shift in proliferation-stimulating activity of DC isolated from TNFR2-deficient mice can not be explained by a clear shift of expression of activation markers on the DC (as shown in Table 2). In contrast to TNFR2-deficient DC, CLP hardly affected the supportive activity of WT DC for CD4 T cell proliferation.
TNF production in healthy mice
As splenic DC of TNFR2-deficient mice resembled DC from post-septic WT mice in terms of IL-12 production capacity, we searched for the common factor shared between septic WT mice and naïve TNFR2-deficient mice. As TNF is strongly increased during sepsis, we wondered whether endogenous bioactive TNF levels might be increased in naïve TNFR2-deficient mice owing to the lack of scavenger-soluble TNFR2. This idea was supported by our recently reported finding of high levels (10–20 ng/ml) of soluble TNFR2 secreted in the urine of naïve WT mice.
22
In order to determine whether TNF is endogenously produced in WT mice, TNF mRNA expression was measured in different organs of naïve WT mice using TNF-deficient mice as control. The results clearly demonstrated that TNF is expressed in naïve WT mice in a number of tissues (Figure 2a); TNF mRNA was below detection level when isolated from the respective organs from TNF-deficient mice. However, owing to the technical problem of detection of low amounts of protein in urine, we were not able to directly detect endogenous TNF in serum or urine, neither in free form nor bound to TNFR2.
(a) mRNA from indicated tissues of naïve WT mice was analyzed for TNF expression. (b) WT mice were pre-treated (pre-treat.) with LPS, TNF or PBS as control and challenged with LPS 24 h later. Ninety min after challenge, spleens were harvested and IL-12-producing cells were determined. (c–e) WT and TNFR2-deficient mice were pre-treated with P. acnes or PBS as a control and challenged with LPS 7 d later (PBS-treated WT: filled circles; P. acnes-treated WT: open circles; PBS-treated TNFR2-deficient: filled squares; P. acnes-treated TNFR2-deficient mice: open squares). Four h after challenge, spleen masses (a), total cell count in spleens (b) and IFN-γ levels in serum were determined. (a–e) Each symbol represents a mouse, line indicates mean. *Statistically significant difference.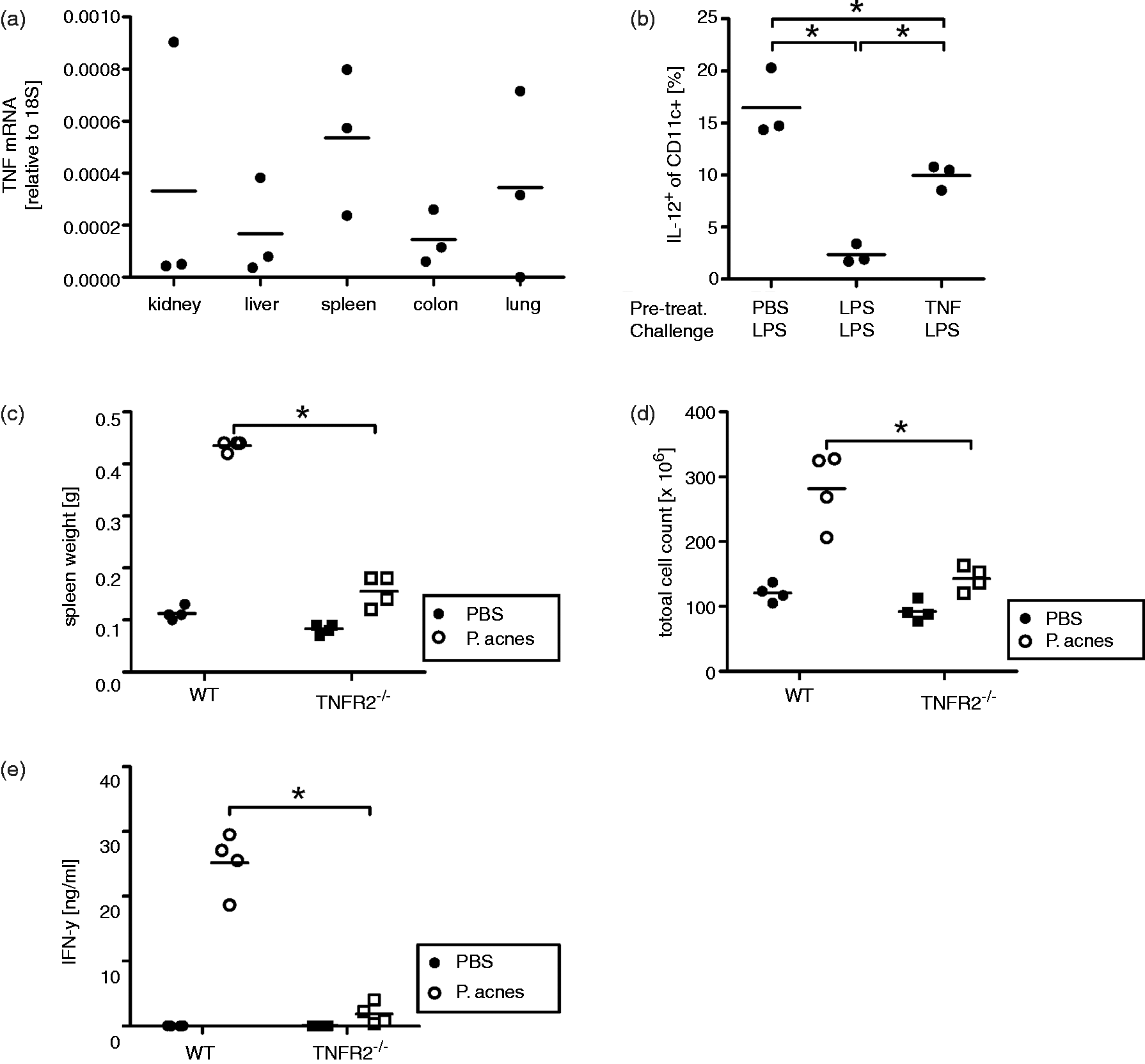
TNF induces habituation in DC
In order to find out whether exposure to TNF is able to induce a state of tolerance in DC, WT mice were tested in the model of LPS tolerance. 23 Similar to the changes in DC function observed after CLP, in vivo pre-treatment with LPS before LPS challenge reduced the proportion of IL-12-producing splenic DC (Figure 2b). 24 To validate TNF as an effector molecule in habituation of DC function, we pre-treated WT mice with TNF before LPS challenge. Similar to LPS pre-treatment, TNF pre-treatment also reduced the levels of IL-12-producing DC upon LPS challenge (Figure 2b).
DC from TNFR2-deficient mice are refractory to P. acnes-induced hyper-sensitization
Because TNFR2-deficient mice mounted mitigated inflammatory responses compared to WT mice after CLP, we assumed that hyper-sensitization to LPS might also be moderated in these mice. Therefore, we subjected TNFR2-deficient mice to the well-established model for induction of LPS hyper-sensitivity by P. acnes injection.25,26 As expected, compared with WT mice sensitized with P. acnes, TNFR2-deficient mice displayed a significantly smaller splenomegaly upon LPS challenge (Figure 2c, d). IFN-γ serum levels were also significantly lower in TNFR2-deficient mice than in WT mice (Figure 2e). Thus, TNFR2-deficient mice were not hyper-sensitized by P. acnes as strongly as WT mice.
Discussion
So far, no clear picture of TNFR2 functions has emerged from the use of mice with a genetically ablated TNFR2 gene. For example, TNFR2-deficient mice have been described to be protected from TNF-induced death and tissue necrosis, 14 from neurovascular lesions in experimental cerebral malaria,27,28 and from experimentally-induced glomerulonephritis. 29 Cooperative effects of both TNF receptors seemed to aggravate TNF-induced arthritis 11 and liver toxicity in experimental hepatitis. 30 In support of these findings, mice over-expressing human TNFR2 displayed increased pathology. 31 Thus, activation of TNFR2 clearly seemed to contribute to inflammation-induced tissue damage. However, in other models, such as in retinal ischemia 32 and pulmonary edema, 33 the lack of TNFR2 exacerbated pathological effects, and selective activation of TNFR2 had a protective effect in oxidative stress-induced neurodegeneration. 34 Ambiguous functions of TNFR2 were seen in the formation and maintenance of granuloma in mycobacterial infections 35 and in experimental autoimmune encephalomyelitis where TNFR1 appeared to be required for the initiation of pathology, while TNFR2 was found to exacerbate the disease.5,36 Previous results from our group showed that TNFR2-deficient mice were neither protected from septic death by pre-treatment with LPS 37 nor did they develop immunoparalysis in sepsis 2 d after induction by CLP. 12 These observations pointed to a regulatory role of TNFR2 affecting the immune status.
In this report we present data which show that a mechanism of TNF-induced habituation to inflammatory stimuli seems to operate in TNFR2-deficient mice at the level of myeloid cells. DC from naïve TNFR2-deficient mice resembled DC of septic WT mice: septic mice have reduced numbers of functionally impaired DC,18,20,38,39 possibly owing to epigenetic regulation. 21 However, the functionally impaired phenotype of primary DC from naïve TNFR2-deficient mice did not reflect epigenetic modification as TNFR2-deficient BMDC and splenic DC or BMDC from chimeric WT mice with a TNFR2-deficient hematopoietic system were not functionally impaired. Thus, our findings confirm previous reports, i.e. DC isolated from septic mice had an impaired capacity for IL-12 production, 40 while in vitro-generated DC from such septic mice did not exhibit diminished IL-12 production but developed into regulatory DC with impaired capacity for Th1 priming when injected into mice. 41 Thus, the impaired function of DC isolated from spleens of TNFR2-deficient mice does not seem to be a direct effect of epigenetic alterations in bone marrow DC precursors. Lack of TNFR2 signaling could be a possible cause for the impaired phenotype. In support of this idea was the observation that TNFR2 seemed to provide a survival signal for DC when they were generated in vitro from bone marrow cells of TNFR2-deficient mice. 42 However, we show that the phenotype of DC from naïve TNFR2-deficient mice resembled DC from septic WT mice. Lack of TNFR2 signaling cannot be the reason for the impaired IL-12 response in sepsis as TNFR2 is strongly induced in inflammatory settings. Furthermore, in the well-established model of LPS-tolerance, naïve WT mice pre-treated with LPS responded in a similar fashion as WT mice 2 d after CLP with reduced IL-12 production. As both sepsis and LPS pre-treatment cause release of TNF, we investigated the impact of TNF pre-treatment on WT mice. Indeed, WT mice pre-treated with TNF demonstrated a phenotype similar to LPS pre-treated or septic mice with a prominent reduction in IL-12-producing DC.
As we found that TNF induced a refractory state in DC, we tested whether endogenous TNF is present in healthy mice. As direct identification of TNF from urine and serum was below the detection limit, TNF mRNA expression was measured and found in multiple organs. Others have previously shown that endogenous TNF in naïve mice is necessary for the homeostatic development of T cells without any on-going immune reaction, which clearly demonstrates that biologically active TNF is continuously present in healthy animals. 43 Together with our previous finding of endogenous soluble TNFR2 secreted with urine from healthy WT mice, 22 it is plausible that endogenous TNF is constantly sequestered bound to soluble TNFR2. Hence, mice deficient in TNFR2 may continuously or repeatedly become exposed to biologically active, non-sequestered endogenous TNF, thereby training the DC. The training seems to cause a shift in the generation/recruitment of immature myeloid cells (MDSC) as seen in sepsis (Table 1), confounding the maturation of DC in response to inflammatory stimuli. This habituation may also explain the refractoriness of TNFR2-deficient mice to hyper-sensitization by P. acnes, in particular, as it has been shown that the IFN-γ response to P. acnes causing hyper-sensitization critically depends on IL-12. 44
It is important to note that DC from TNFR2-deficient mice were not completely non-functional. DC from naïve TNFR2-deficient mice induced stronger T cell proliferation than DC obtained from naïve WT mice even though no enhanced expression of activation markers was measured on TNFR2-deficient DC (Table 2). This observation and the fact that CLP induced a clear inversion of the proliferation-stimulating activity of DC isolated from the two mouse lines demonstrate that additional mechanisms such as regulatory cells might be at work for the manifestation of in vivo suppression in sepsis. Interestingly, the appearance of regulatory T cells in sepsis was impaired in TNFR2-deficient mice. 13 The differential reaction of WT and TNFR2-deficient mice to CLP-induced sepsis strongly reducing the numbers of inflammatory myeloid cells in septic WT mice, whereas no change was induced in TNFR2-derived inflammatory myeloid cell numbers points to a role of these potentially suppressive immature myeloid cells in the habituated phenotype observed with TNFR2-deficient mice. However, whether TNFR2 signaling plays a role in cells participating in this interaction remains to be analyzed.
In conclusion, TNF exposure habituates DC leading to reduced IL-12 responses. This effect is prevented by TNFR2 in naïve mice, but emerges after exposure to high levels of TNF during inflammation or when endogenous TNFR2 is missing. As we have recently shown in the model of immune complex-induced glomerulonephritis, 22 this training of DC leads to adapted responses following inflammatory reactions. Obviously, previous exposure to endogenous TNF is capable of conferring such a suppression-inducing phenotype on DC in TNFR2-deficient mice. 12 This calls for a careful interpretation of data obtained with TNFR2-deficient mice as training by endogenous TNF and not (only) lack of TNFR2 signal transduction will impact on the results in such experimental models.
Footnotes
Funding
This work was supported by a German Research Foundation (DFG) grant Ma760/19-1 (to DNM).