
Abstract
We used the LEW1.WR1 model of Kilham rat virus (KRV)-induced type 1 diabetes (T1D) to test the hypothesis that blocking IL-1 pathways early in the course of the disease can modulate virus-induced innate immunity and prevent disease progression. Administering KRV plus IL-1 receptor antagonist (Anakinra) for 14 d prevented insulitis and T1D. Anakinra reversed the KRV-induced systemic inflammation evidenced by the accumulation of T cells in the spleen and pancreatic lymph nodes on d 5 post-infection. Blocking IL-1 modulated the level of IRF-7 and IL-6 gene expression in the spleen and the p40 subunit of IL-12 and IL-23 in the serum. Anakinra did not interfere with the ability of LEW1.WR1 rats to clear the virus from the spleen, pancreatic lymph nodes or serum. Consistent with these data, normal levels of KRV-specific adaptive immune responses were detected in in the spleen and peripheral blood of the treated animals. Finally, blocking IL-1 pathways reversed the KRV-induced modulation of gut bacterial communities. The data may imply that IL-1 pathways are directly linked with early mechanisms whereby KRV infection leads to islet destruction, raising the hypothesis that blocking IL-1 pathways early in the course of the disease could be a useful therapeutic approach for disease prevention.
Keywords
Introduction
Type 1 diabetes (T1D) is a pro-inflammatory autoimmune disease that primarily affects young children and adolescents, causing the slow and progressive destruction of islet beta cells and leading to a complete loss of insulin secretion.1–3 The disease course is typically associated with the appearance of anti-islet auto-Abs in the peripheral blood long before the onset of the disease. 4 How T1D is triggered is unknown, but epidemiological evidence implicates environmental factors, possibly virus infections, in the mechanisms of the disease.5–7
We have used the LEW1.WR1 rat model of T1D to understand mechanisms of virus-induced T1D. This animal has normal levels and functions of T lymphocytes. 8 Infecting LEW1.WR1 rats with Kilham rat virus (KRV) leads to insulitis and T1D 2–4 wks after virus infection. 9 The disease is characterized by a selective loss of islet beta cells, glycosuria, ketonuria and polyuria.8,10
We recently hypothesized that innate immune system activation early after infection plays a major role in the development of virus-induced T1D.9,11,12 Support for this possibility was provided by the finding that virus infection induces a robust pro-inflammatory response in the pancreatic lymph nodes and spleen shortly after virus infection, as evidenced by the up-regulation of IL-1-related pathways in lymphoid organs from infected animals. 13 Furthermore, we recently observed that down-modulation of inflammation with steroids 13 or antibiotic therapy 14 can prevent virus-induced islet destruction.
Emerging data implicate IL-1-related pathways in the mechanism that leads to islet destruction and T1D. 15 IL-1 is expressed in pancreatic islets in the non-obese diabetic mouse early in the course of T1D, and it is also found in the pancreatic lymph nodes and the spleen from LEW1.WR1 rats shortly after KRV infection. 13 In addition, IL-1 synergizes with TNF-α and IFN-γ to induce apoptosis of islet β-cells in vitro. 15 Blocking IL-1-related pathways reduces the incidence of cyclophosphamide-induced T1D in the non-obese diabetic (NOD) mouse. 16
Data from a recent clinical trial suggested that therapy with Anakinra or a mAb against IL-1β did not improve islet function in subjects with newly diagnosed T1D. 17 In this study, we used the LEW1.WR1 rat model to address the possibility that blocking IL-1 pathways early in the course of the disease prior to insulitis or T1D can modulate virus-induced innate immune responses and prevent islet destruction. Our data raise the hypothesis that IL-1 blockade could be a useful therapeutic strategy for diabetes prevention in genetically susceptible individuals.
Materials and methods
Animals and viruses
Specific pathogen-free LEW1.WR1 rats were obtained from BRM (Worcester, MA, USA). Animals were bred and housed in a specific pathogen-free facility and maintained in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences,1996) and the guidelines of the Institutional Animal Care and Use Committee of the University of Colorado Denver. KRV was propagated and titered as previously described. 18
Virus-induced diabetes, IL-1 blockade, blood and lymphoid organ removal
Rats at 21–25 d of age of either sex were injected i.p. with 1 × 107 plaque-forming units (PFU) of KRV as previously described. 19 Anakinra (Amgen, Thousand Oaks, CA, USA) was administered i.p. at a dose of 50 µg/g body mass beginning on the day of infection. To analyze the effect of Anakinra administration on the course of virus-induced T1D, rats were administered KRV only, KRV plus Anakinra, or Anakinra only beginning on the day of virus inoculation. The animals were monitored for insulitis and T1D for 40 d following infection or were sacrificed on d 5 for gene expression and flow cytometry studies. Spleens, pancreatic lymph nodes and blood were collected 5 or 40 d after virus infection. The sera were separated immediately after blood collection and were stored at −80℃ until use. The pancreata were removed for insulitis evaluation 21 and 40 d after virus infection or following disease onset. Cells were obtained from the lymphoid organs by passing the tissue through a 70-µm nylon mesh filter. In the case of the spleen, erythrocytes were lysed with NH4Cl. The cells were washed and re-suspended in tissue culture media or PBS for further use.
Histological staining
Pancreatic tissue was fixed for 24 h in 10% neutral-buffered formalin, embedded in paraffin, cut (5–6 µm slices) and mounted on microscope slides. The tissue was stained with hematoxylin and eosin.
DNA purification and quantitative PCR analysis for the detection of intestinal bacteria
Bacterial DNA was recovered using the QIAmp DNA stool mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. DNA concentrations were determined using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). DNA from Lactobacillus, Bifidobacterium, Clostridium and Bacteroides was detected by quantitative PCR analysis using previously published primers. 20 The data were normalized to the total bacterial DNA in each sample, using recently described primers and conditions. 20
RNA extraction, cDNA synthesis and quantitative RT-PCR
RNA extraction, cDNA synthesis and quantitative RT-PCR were performed as previously described. 21 The primers were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA). Their sequences have been previously published. 21 To determine the KRV transcript levels, we used the following primers: 5′-GGAAACGCTTACTCCGATGA-3′ and antisense 5′-AACCGATGTCCTTCCCATTT-3′.
Flow cytometry
Cells were analyzed by flow cytometry as previously described. 14 A PerCP-conjugated anti-TCRα/β Ab (clone R73, mouse IgG1), a phycoerythrin-conjugated anti-IL-2 R α-chain Ab (CD25,clone OX-39, mouse IgG1), an APC-conjugated anti-CD4 Ab (clone OX-35, mouse IgG2a), a PerCP-conjugated anti-CD8α chain Ab (clone OX-8, mouse IgG1), a FITC-conjugated anti-CD45R Ab (a marker of B cells, clone HIS24, mouse IgG2b) and appropriate isotype controls were purchased from BioLegend (San Diego, CA, USA). An Efluor 450-conjugated mAb against Foxp3 (Clone FJK-16 s, rat IGg2a) and fixation and permeabilization buffers were purchased from eBioscience (San Diego, CA, USA). Flow cytometry was performed using a CYAN ADP instrument (Beckman Coulter; Fullerton, CA, USA), and the results were analyzed with FlowJo software.
KRV-specific cellular and humoral immunity, and serum levels of IL-12 p40
KRV-specific CD8+IFN-γ+ and anti-KRV Abs were detected in the spleen on d 12 after viral infection, as previously described. 11 Anti-KRV Abs were detected in serum samples using a commercially available ELISA kit according to the manufacturer’s instructions (Alpha Diagnostic International, San Antonio, TX, USA). The levels of serum IL-12 p40 were quantified using a kit from Life Technologies (Grand island, NY, USA) according to the manufacturer’s instructions.
Statistical analysis
Statistical comparisons of diabetes-free survival among groups were performed using the method of Kaplan and Meier. Comparisons between more than two groups were performed with a one-way ANOVA with Bonferroni’s multiple comparison test. Comparisons between two groups were performed with an unpaired t-test.
Results
Anakinra prevents insulitis and virus-induced T1D in LEW1.WR1 rats
Our recent studies demonstrated that infection with KRV induces the up-regulation of IL-1 pathways in the pancreatic lymph nodes from LEW1.WR1 rats shortly after virus infection.13,21 We therefore tested the hypothesis that IL-1-related pathways are involved in the mechanism of virus-induced T1D in the LEW1.WR1 model. Animals were injected with 1 × 107 PFU of KRV and treated with Anakinra once a day, beginning on the day of viral inoculation and continuing for 14 or 5 d. The results shown in Figure 1a indicate that infection with KRV resulted in T1D in 60% of the animals (n = 49). In contrast, treating the rats with Anakinra for 14 d reduced the incidence of T1D to 17% (n = 12, P = 0.03 versus animals treated with KRV only). Treating the rats with Anakinra for 5 d did not lead to a difference in the disease incidence compared to KRV only; however, interpretation of this finding is limited due to the small sample size (n = 6).
Kaplan–Meier analysis of KRV-induced T1D in LEW1.WR1 rats treated with Anakinra. LEW1.WR1 rats at 21–25 d of age and of either sex were injected i.p. with KRV. Animals were either injected with KRV only or were injected with KRV and treated with Anakinra for 5 or 14 consecutive d; control animals were left untreated. Animals were tested for T1D for 40 d after viral inoculation. Diabetes was defined as having plasma Glc concentrations > 250 mg/dl (11.1 mmol/l) on two consecutive days. Diabetes-free survival was analyzed using the Kaplan–Meier method (a). Statistical analyses were performed using the log rank test. Shown are paraffin sections of hematoxylin and eosin-stained sections of pancreata removed from control LEW1.WR1 rats that were left untreated (uninfected), KRV-infected rats at 21 d following infection (insulitis), KRV plus Anakinra-treated animals at 40 d following infection, and KRV-infected rats at diabetes onset (T1D) (b).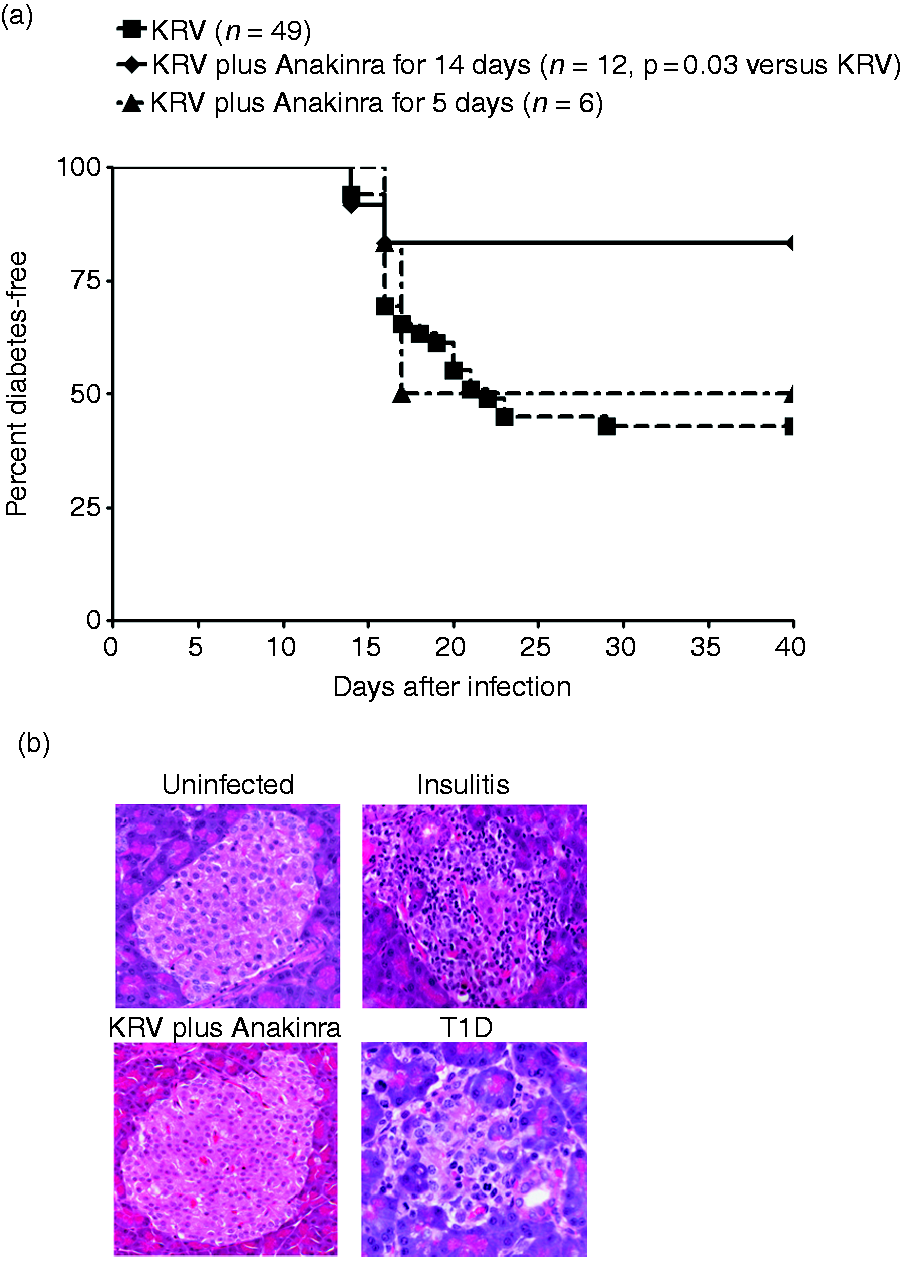
We next determined whether Anakinra-induced disease prevention was associated with altered insulitis (n = 6 per group; ≥ 20 islets per animal were screened for insulitis). We observed that the pancreata from KRV-infected rats showed insulitis in the majority of the islets on d 21 after virus inoculation, with severe islet destruction observed following disease onset (Figure 1b). In contrast, the pancreata from rats infected with KRV and treated with Anakinra did not show islet infiltration or destruction (Figure 1b). Collectively, these data suggest that Anakinra protects LEW1.WR1 rats from islet infiltration and T1D, thus suggesting that IL-1 pathways are directly involved in the virus-induced islet destruction in the LEW1.WR1 model.
Anakinra restores KRV-induced accumulation of T and B cells in the spleen and pancreatic lymph nodes
We have previously shown that infection with KRV induces the accumulation of T cells in the spleen and pancreatic lymph nodes on d 5 post-infection, and we hypothesized that this effect could be involved in the mechanism of virus-induced T1D.13,18,22 We addressed the possibility that Anakinra prevents T1D by reversing virus-induced alterations in the T and B cell compartments. We were particularly interested in determining whether the mechanism of Anakinra-induced disease amelioration involves changes in the frequency or number of CD4+Foxp3+Treg cells in the spleen or pancreatic lymph nodes. To test our hypothesis, animals were left untreated (n = 7–16), were infected with KRV (n = 3–11), were infected with KRV and treated with Anakinra (n = 8–9), or were treated with Anakinra only (n = 3) beginning on the day of infection. Spleens and pancreatic lymph nodes were harvested 5 d after virus infection, and the cells were counted and analyzed by flow cytometry for the expression of CD4, CD8, TcRαβ, CD45R (a marker of B lymphocytes in the rat) and the co-expression of CD4 and Foxp3. The data presented in Figure 2a indicate that KRV induced an increase in the absolute numbers of CD4+, CD8+ and TcRαβ+ cells, and a slight reduction in the proportion of B cells in the spleen. However, treating infected animals with Anakinra for 5 d significantly reduced the absolute number of CD4+, CD8+ and TcRαβ+ cells compared with KRV only. In contrast, the absolute number of Treg cells in the spleen from rats treated with KRV plus Anakinra was similar to that found in animals administered with KRV only.
Numbers of lymphocyte subsets in the spleen and pancreatic lymph nodes of Anakinra-treated rats. LEW1.WR1 rats were left untreated, were infected with KRV, were injected with KRV plus Anakinra beginning on the day of infection or were treated with Anakinra only. Cells from the spleen and pancreatic lymph nodes were harvested on d 5 post-infection, counted and stained for flow cytometry analysis with fluorochrome-conjugated mAbs directed against the indicated lymphocyte marker. The scatter dot plots represent cell numbers in the spleen (a) and pancreatic lymph nodes (b) from individual rats. Statistical analyses were performed using an ANOVA with Bonferroni's correction for multiple comparisons. *P < 0.001; **P < 0.01; ***P < 0.05.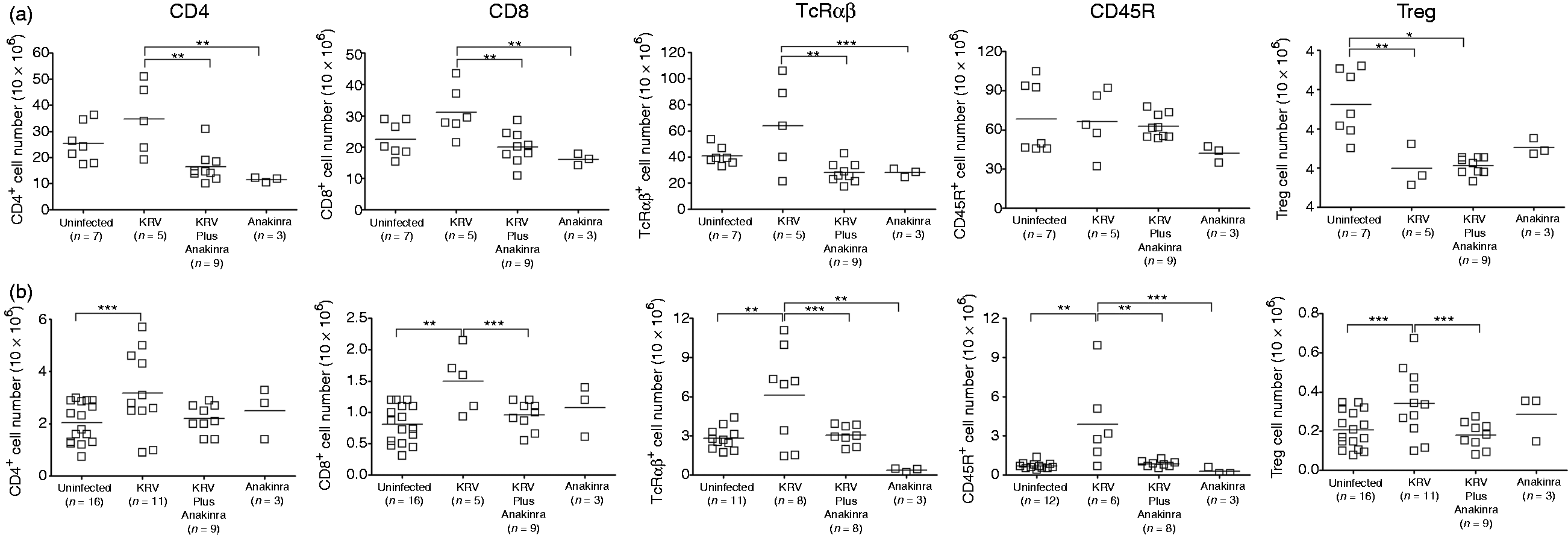
Treating animals with KRV resulted in an increase in the absolute number of T and B cells compared with the uninfected control in pancreatic lymph nodes (Figure 2b). Treating rats with KRV plus Anakinra reversed the KRV-induced alterations, resulting in diminished cell numbers when compared with KRV only. Taken together, the data may suggest that IL-1 blockade reverses the virus-induced accumulation of T and B lymphocytes in the spleen or pancreatic lymph nodes. The data also suggest that Anakinra-induced disease prevention is unlikely to involve the up-regulation of Treg cells in the spleen or pancreatic lymph nodes on d 5 post-infection.
Anakinra modulates KRV-induced gene expression in the spleen
We tested the hypothesis that Anakinra could prevent islet destruction via a mechanism that may involve intervening with KRV-induced innate up-regulation in the spleen.13,21,23 To investigate this possibility, we evaluated the levels of the transcripts for IRF-7, CXCL-10 and IL-6 in the spleen from naïve uninfected rats (n = 5–6), rats administered KRV only (n = 10–12), rats administered with KRV plus Anakinra (n = 6–7) or rats treated with Anakinra only (n = 5–6) on d 5 post-infection. We also assessed the level of transcripts for KRV on d 5 post-infection. The expression of transcripts for KRV is an indicator of viral replication.24,25 The results shown in Figure 3 demonstrate that transcripts for KRV were detectable in the spleen from KRV- and KRV-plus-Anakinra-treated animals, but not in the spleen from naïve rats or animals treated with Anakinra only. The data further indicate that KRV infection induced a significant increase in the level of transcripts for IRF-7 and CXCL-10 (P < 0.01 for both genes). However, Anakinra administration reduced the level of IRF-7 and increased the level of IL-6 (P < 0.05 for both genes). The splenic level of IL-6 was also increased in rats treated with Anakinra only compared with KRV only (P < 0.001) or the uninfected control (P < 0.01). We did not observe any differences in the level of KRV-induced IL-1β gene expression in the spleen from KRV plus Anakinra-treated rats compared with KRV only-treated rats. In addition, no differences in the expression levels of the genes that were studied in the spleen were found in the pancreatic lymph nodes from rats treated with KRV plus Anakinra compared with KRV only. Taken together, these data could imply that Anakinra-induced disease prevention may be linked with the modulation of virus-induced gene expression in the spleen shortly after infection.
KRV and pro-inflammatory gene expression in the spleen of Anakinra-treated rats. LEW1.WR1 rats were left untreated, were injected with KRV with or without Anakinra treatment beginning on the day of infection, or were administered with Anakinra only, as shown. RNA was extracted from the spleen 5 d post-infection, and the expression levels of the indicated genes were assessed using quantitative RT-PCR. The results are presented as the mRNA expression of the gene of interest relative to the expression of β-actin in tissue from individual animals. Statistical analyses were performed using an ANOVA with Bonferroni's correction for multiple comparisons. *P < 0.001; **P < 0.01; ***P < 0.05.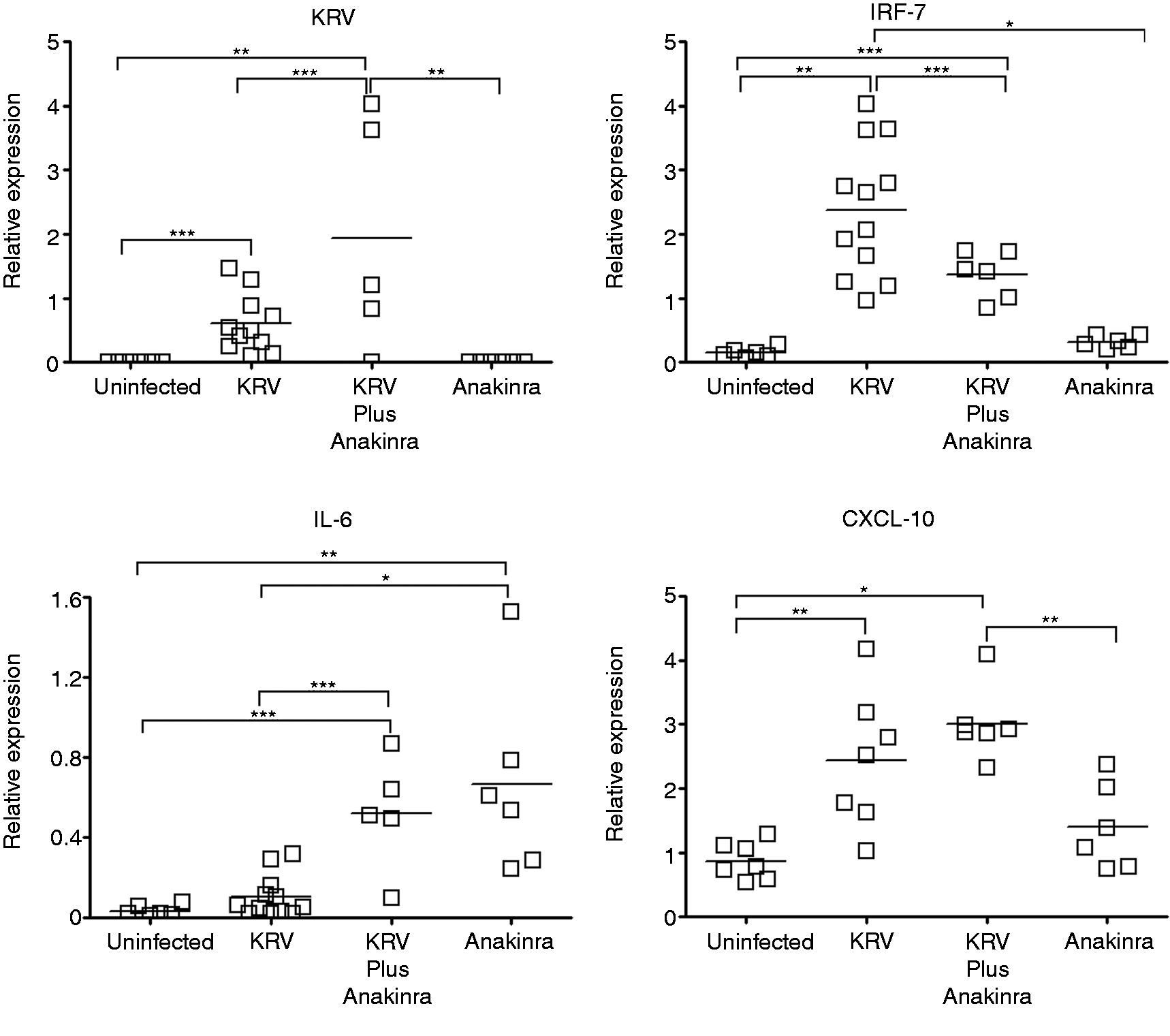
Anakinra lowers KRV-induced p40 levels in the peripheral blood
We next tested the hypothesis that Anakinra-induced disease prevention may be linked to altered serum expression levels of the p40 subunit of IL-12 and IL-23. To that end, LEW1.WR1 rats were left untreated (n = 3),were infected with 1 × 107 PFU of KRV (n = 5), were injected with KRV plus Anakinra for 5 consecutive d beginning on the day of infection (n = 5) or were administered with Anakinra only (n = 3). Sera were recovered on d 5 following the virus infection. The data shown in Figure 4 demonstrate that rats infected with KRV experienced a 10-fold increase in the expression level of the p40 subunit in the serum compared with uninfected rats (P < 0.001). However, the administration of KRV plus Anakinra led to a 25% reduction in the level of the p40 subunit (P < 0.05 compared with KRV only). Injection of Anakinra only did not alter the p40 levels compared with those in the uninfected controls. These observations suggest that blocking IL-1 pathways interferes with KRV-induced expression of IL-12 or IL-23 in the serum.
Expression levels of the p40 subunit in serum from Anakinra-treated rats. Groups of LEW1.WR1 rats 21–25 d of age were left untreated, infected i.p. with KRV, treated with KRV plus Anakinra or injected with Anakinra only. Sera were recovered on d 5 post-infection, and the serum levels of the p40 subunit of IL-12/IL-23 were assessed by ELISA. Shown is a scatter dot plot representing the p40 levels in the sera from individual rats. Statistical analyses were performed using an ANOVA with Bonferroni's correction for multiple comparisons. *P < 0.001; ***P < 0.05.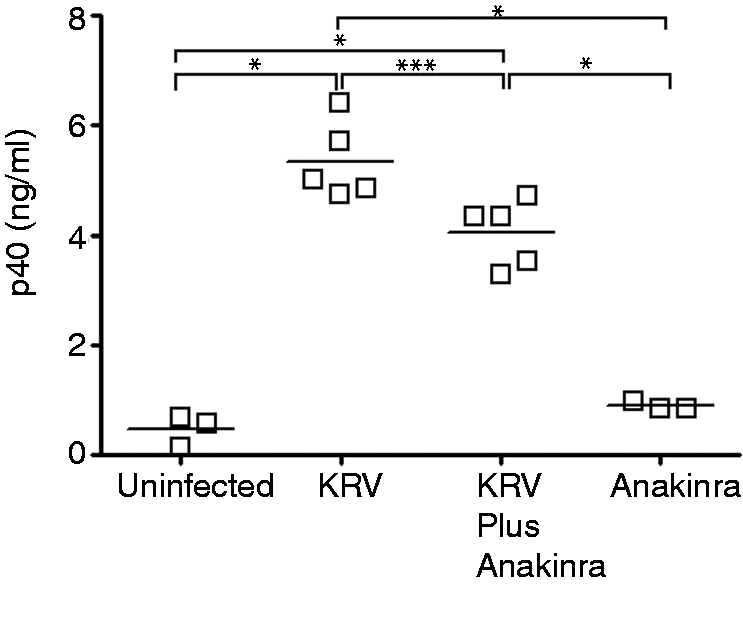
Anakinra does not interfere with the induction of KRV-specific cellular and humoral immunity and virus clearance
Given the observation that Anakinra treatments altered the innate immune responses in the spleen and serum, we addressed the possibility that IL-1 blockade interferes with virus-specific adaptive immune responses. For this purpose, we analyzed the effect of Anakinra injection on the proportion of KRV-specific T cells in the spleen 12 d after virus infection. Rats were left untreated (n = 3), were infected with KRV only (n = 12), were administered with KRV plus Anakinra for 12 d beginning on the day of infection (n = 5) or were treated with Anakinra only for 12 d (n = 3). The data presented in Figure 5a show that treating rats with KRV plus Anakinra did not lead to differences in the percentage of CD8+IFN-γ+ cells compared with rats treated with KRV only. Likewise, data shown in Figure 5b show that IL-1 blockade did not alter the levels of KRV-specific Abs present in the serum on d 14 and 40 (n = 3 per group).
KRV-induced humoral and cellular immunity and virus clearance. LEW1.WR1 rats were left untreated, were infected with KRV, were treated with KRV plus Anakinra or were injected with Anakinra only. For KRV-specific T-cell responses, spleen cells were harvested on d 12 post-infection. Spleen cells were surface-stained with a mAb against CD8 and labeled intracellularly with a mAb against IFN-γ (a). To analyze the KRV-specific Ab responses, sera were collected 14 and 40 d post-infection. For addressing KRV clearance, transcript levels were quantitated in the spleens, pancreatic lymph nodes, and sera on d 5 or 40 following virus inoculation (c). The results are presented as the expression of the KRV gene mRNA relative to the expression of β-actin. Bars represent mean values. Statistical analyses between multiple groups were performed using an ANOVA with Bonferroni's correction for multiple comparisons. *P < 0.001; **P < 0.01; ***P < 0.05.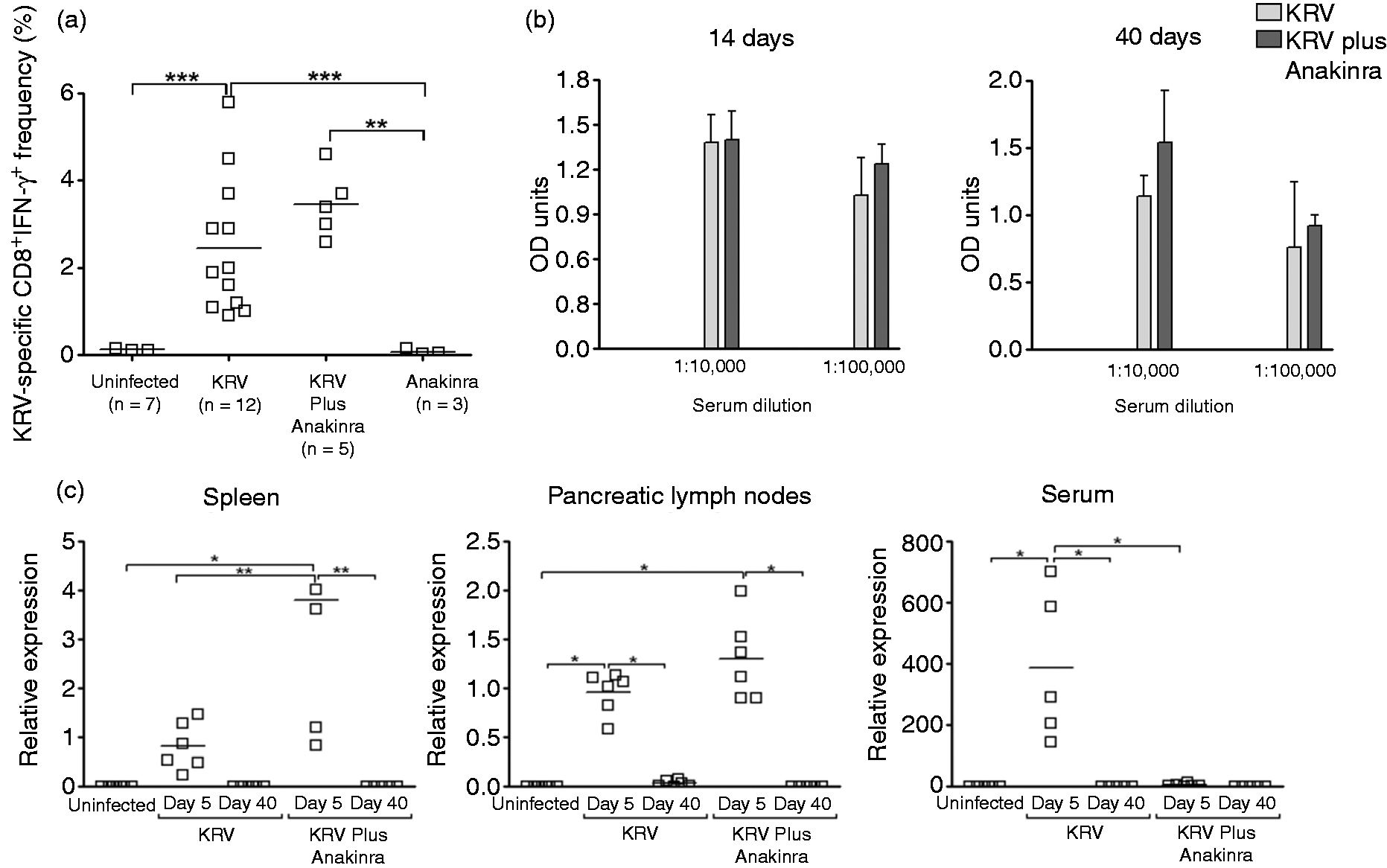
We next used quantitative RT-PCR to analyze the effect of Anakinra on the ability of the rats to eradicate the virus from the spleen, pancreatic lymph nodes, and serum (n = 4–6 per group). Infection with KRV combined with Anakinra resulted in a significant increase in the level of transcripts for KRV in the spleen on d 5 post-infection (Figure 5c). Unlike the spleen and pancreatic lymph nodes, blocking IL-1 pathways resulted in remarkably reduced levels of transcripts for KRV in the serum. Lastly, the amount of KRV transcripts in the spleen, pancreatic lymph nodes and serum on d 40 post-infection was below the detection limit, as was observed in control uninfected rats. Taken together, the data suggest that blocking IL-1 does not influence the magnitude of KRV-specific T-cell and Ab responses or virus elimination.
IL-1 blockade reverses virus-induced altered intestinal microbiome
We recently proposed that interplay between virus-induced inflammation and the intestinal microbiome may be involved in the early course of T1D in the LEW1.WR1 rat model.14,22 Here, we examined the effect of IL-1 blockade on the gut microbiota from infected rats. To that end, animals were left untreated, administered KRV with or without Anakinra beginning on the day of infection, or treated with Anakinra only (n = 5–6 per group). Fecal samples were collected on d 5, a time point at which alterations in the intestinal microbiota are detectable, and on d 12, a time point at which these differences are not observed.
14
We assessed the abundance of bacterial genera that we previously found to be elevated (Bifidobacterium spp. and Clostridium spp.) or to be unchanged (Bacteroides spp. and Lactobacillus spp.) shortly after virus infection.14,22 The data presented in Figure 6a show that, as previously seen, KRV infection significantly increased the abundance of the Bifidobacterium spp. and Clostridium spp. genera compared with untreated animals (P < 0.01 and P < 0.05, respectively). However, treating animals with KRV plus Anakinra significantly reduced the abundance of both of these genera (P < 0.01 and P < 0.05, respectively, compared with KRV only). On d 12 post-infection, no significant differences were detected in the abundance of the bacteria tested in fecal samples from rats treated with KRV plus Anakinra compared with KRV only (Figure 6b). These data imply that IL-1 pathways may be involved in the mechanism in which KRV modulates the intestinal microbiome.
The gut microbiome in Anakinra-treated animals. Animals were left untreated, were administered KRV with or without treatment with Anakinra beginning on the day of viral infection, or were treated with Anakinra only. DNA was extracted from fecal samples collected from individual rats on d 5 (a) and 12 (b) post-infection. Quantitative PCR was used to assess the level of various bacterial genera, as indicated in the figure. Standard curves were obtained using DNA extracted from a fecal sample from a B6 mouse. Shown are relative DNA levels that were calculated using a bacterial reference gene. Statistical analyses were performed using ANOVA with Bonferroni's correction for multiple comparisons. *P < 0.001; **P < 0.01; ***P < 0.05.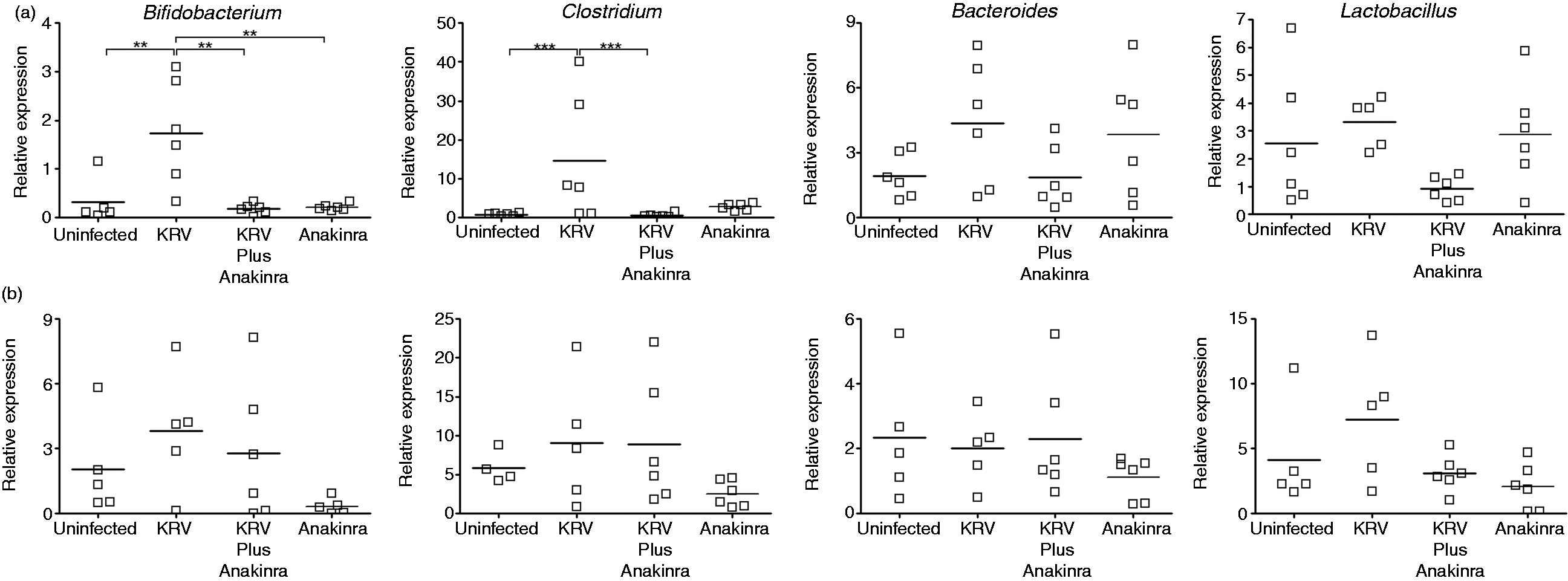
Discussion
IL-1 is a pro-inflammatory cytokine that stimulates both local and systemic responses (reviewed in Gabay et al. 26 ). IL-1 facilitates the recruitment of inflammatory cells to the site of inflammation by inducing the up-regulation of adhesion molecules on endothelial cells, the release of chemokines by stromal cells, and the production of acute-phase proteins. 26 Here, we used the LEW1.WR1 rat model of virus-induced T1D to test the hypothesis that IL-1 blockade with IL-1RA can prevent virus-induced immune pathology in islet beta cells. Our data demonstrate for the first time that blocking IL-1 pathways beginning early in the course of T1D can prevent islet autoimmunity triggered by virus infection. We show that IL-1 blockade modulates the KRV-induced inflammatory responses without intervening in the virus-specific adaptive immune responses or viral clearance. The observation that a 5-d treatment with Anakinra was not sufficient to prevent T1D may suggest that IL-1 pathways are involved in the early- and late-phase of disease development. We hypothesize that blocking IL-1 pathways early during the course of diabetes may prove to be an effective therapeutic approach for disease prevention.
The data from the present study indicate that IL-1 blockade reverses the virus-induced systemic inflammation reflected by the accumulation of T cells in the spleen and pancreatic lymph nodes shortly after infection, 13 implicating IL-1 pathways in the mechanism of this virus-induced systemic effect. We postulate that the decrease in inflammation may reduce the pool of potentially autoreactive T cells, thus sparing pancreatic islets from full-blown autoimmune attack and islet destruction. Our findings are reminiscent of those of a previous study that indicated that blocking the IL-1β pathway in a mouse model of virus-induced chronic intestinal inflammation reversed the virus-induced up-regulation of pro-inflammatory molecules and systemic inflammation. 27
The data indicating that IL-1 pathways are involved in the mechanism of virus-induced T1D in the LEW1.WR1 model are compatible with our other recent observations from the BBDR and the LEW1.WR1 rat models of virus-induced T1D.13,21 We reported that infecting these animals with KRV induces a robust activation of the innate immune system in the spleen and pancreatic lymph nodes, which was reflected by a substantial increase in the gene expression levels of IL-1RA, IL-1α, IL-1β and other pro-inflammatory molecules on d 5 post-infection. 21 Because Anakinra blocks the receptor for both IL-1α and IL-1β, 26 the role that each of these cytokines plays in the development of T1D remains to be elucidated. IL-1α is expressed under normal physiological conditions, but under some pathological conditions, such as ischemia, it is translocated to the cell surface and activates other cells to trigger sterile inflammation. 28 IL-1β is expressed by macrophages and dendritic cells, and can promote inflammation and autoreactive T cell activation in the periphery and, subsequently, around islet beta cells.26,29,30As mentioned earlier, IL-1 pathways are linked with the induction of adhesion molecules on mesenchymal cells and endothelial cells. Thus, together with chemokines, these pathways may facilitate the infiltration of innate and adaptive immune cells from the circulation into the target tissue. 30
Whether and how IL-1 pathways are involved in the mechanism of human T1D is currently unclear. Our recent reports,31,32 and data from other laboratories, have linked IL-1 cytokine family members, including IL-1β, IL-1R1 and IL-1R2 to T1D in humans. 33 Recent clinical trials conducted in participants after disease diagnosis indicated that IL-1 blockade with Anakinra or an anti-IL-1β Ab did not have a beneficial effect on islet function. 17 One potential explanation for these observations could be the timing of the therapy, which was given at a late disease stage after disease onset. IL-1 blockade was recently shown to be clinically effective in pro-inflammatory conditions, such as type 2 diabetes, rheumatoid arthritis and systemic juvenile idiopathic arthritis. 28
Studies addressing the role of IL-1-related pathways in the development of T1D in the NOD mouse led to mixed results. NOD mice injected with soluble IL-1 R were protected from islet destruction, but not from insulitis in a dose-dependent manner via mechanisms that did not involve altered adaptive immune responses. 16 In addition, recent studies indicated that a combination of anti-CD3 mAb with IL-1RA is synergistic in reversal of T1D. 34 Yet, other reports suggested that NOD mice with disrupted IL-1 R expression 35 or a deficiency in caspase-1, 36 an enzyme required for the expression of mature IL-1β, developed disease with a similar incidence as control mice. The data from NOD mice with disrupted IL-1 signaling may suggest that IL-1-related pathways are not involved in disease development; however, it could also be that redundant pathways in these genetically manipulated mice compensated for the disrupted IL-1 pathways and permitted the development of T1D. 15
Our data suggest that the mechanism whereby IL-1 blockade prevents islet destruction is unlikely to involve altered virus-induced humoral and cellular immunity. This hypothesis is in agreement with an earlier report demonstrating that NOD mice treated with IL-1RA had a normal distribution of mononuclear cell subsets, Con A-induced T cell activation, and expression of IFN-γ and IL-2. 16 Rather, our findings may suggest that the mechanism by which the disease is ameliorated may involve altered innate immune responses, as evidenced by the down- and up-regulation of IRF-7 and IL-6, respectively, in the spleen, and by the diminished level of the p40 subunit in the serum. The mechanism leading to the elevated level of IL-6 transcripts in the spleen of Anakinra-treated rats is unknown. The regulation of IL-6 gene expression occurs mainly at the transcriptional level and therefore its overexpression may be linked with the up-regulation of NF-IL6 and NF-κB, transcription factors involved in IL-6 synthesis, 37 or down-regulation of negative regulators, such microRNAs. 38 Reduced levels of IRF-7 and IL-12 (or IL-23) and an increased level of IL-6, a pleiotropic cytokine with both inflammatory and anti-inflammatory properties, 39 could potentially lead to the attenuation of islet-specific adaptive immune responses and subsequent disease amelioration.12,40 Earlier reports indicated that IL-6 promotes Th2 differentiation and simultaneously inhibits Th1 polarization. 41 Given the fact that IL-1β induces itself, 30 it was surprising to find that the level of transcripts for IL-1β in Anakinra-treated rats was unaffected (data not shown). This unexpected observation may be explained by the fact that under some circumstances IL-1RA can act both as an inhibitor and positive regulator of IL-1 expression. 42
One other potential mechanism by which Anakinra could prevent the death of islets is by blocking the IL-1 receptor expressed by beta cells. Pancreatic islets are highly sensitive to IL-1, more so than other tissues, presumably because they express the highest density of IL-1 receptors out of all body tissues. 43 Indeed, IL-1 alone or in combination with other pro-inflammatory cytokines, such as type I IFNs can induce beta cell destruction in islets from humans and animals and in perfused pancreas. This destruction occurs via pathways involving MAPK and NF-κB.44–46In addition, animals not normally susceptible to diabetes that were injected with IL-1 developed transient insulinopenic diabetes. 46 It is possible that Anakinra binds to the IL-1 receptor expressed by beta cells, thus competing with the binding of IL-1 and preventing the recruitment of the IL-1 receptor accessory protein that is required for delivery of the signal. 15 Experiments are underway to better understand the role of pancreatic islets in the mechanism of Anakinra-induced disease prevention.
We recently hypothesized that cross-talk between the innate immune system and intestinal bacteria shortly after infection may play a key role in disease mechanisms. 14 Our data, which indicate that Anakinra restored normal levels of the Bifidobacterium and Clostridium genera on d 5 post-infection, are compatible with the hypothesis that IL-1 pathways maybe involved in triggering viral-induced modulation of the intestinal microbiota. Whether changes in the abundance of gut bacterial communities are linked with mechanisms of autoimmunity remain to be determined. In any case, these observations raise the possibility that changes in gut bacterial communities could be used as a sensitive marker for monitoring inflammation and autoimmunity.
In summary, our findings suggest that blocking IL-1- pathways early in the course of T1D could be a useful therapeutic approach to prevent islet destruction. Additional studies performed in subjects at high risk for the development of T1D prior to disease diagnosis will be required to determine whether blocking IL-1 can protect genetically susceptible individuals from islet destruction.
Footnotes
Funding
This study was supported by grants 1-2006-745, 1-2007-584, 5-2008-224, and 5-2011-41 from JDRF (to DZ).