
Abstract
β-Arrestins 1 and 2 couple to seven trans-membrane receptors and regulate G protein-dependent signaling, receptor endocytosis and ubiquitylation. Recent studies have uncovered several unanticipated functions of β-arrestins, suggesting that the role of β-arrestins in cell signaling is much broader than originally thought. It is now recognized that β-arrestins can transduce receptor signaling independent of G proteins. The expression of β-arrestins is differentially regulated in immune cells and tissues in response to specific inflammatory stimuli, and β-arrestins are critical regulators of the inflammatory response. This review will focus on β-arrestins in immune cells and the impact of altered expression on the pathogenesis of specific inflammatory diseases. Understanding the role of β-arrestins in inflammation may lead to new strategies to treat inflammatory diseases, such as sepsis, rheumatoid arthritis, asthma, multiple sclerosis, inflammatory bowel disease and atherosclerosis.
β-Arrestins mediate receptor endocytosis, ubiquitylation and G protein-dependent and G protein-independent signaling
The seven trans-membrane receptors (7-TMRs), also referred to as G protein-coupled receptors (GPCRs), are the largest class of cell surface receptors in the human genome. The 7-TMRs have immense therapeutic importance and regulate diverse physiological and pathophysiological processes. 1 Ligand binding to 7-TMRs activate heterotrimeric G proteins that transiently drive second messengers (e.g. cAMP, inositol triphosphate and intracellular Ca2+) and initiates intracellular signals. 2 β-Arrestins, which are non-visual arrestins, are referred to as β-arrestin 1 and β-arrestin 2. β-Arrestins were initially characterized in the context of β-adrenergic receptor signaling as ‘arresting’ proteins, which desensitize agonist-induced 7-TMR signaling. 3 Following agonist binding to 7-TMRs, the receptors are phosphorylated at the carboxyl terminus or intracellular loops by GPCR kinases. 4 Phosphorylation promotes recruitment of β-arrestins to 7-TMRs, which, in turn, serve as a scaffolding protein. Clathrin and beta 2-adaptin are recruited to the plasma membrane β-arrestin complex and initiate inward endocytosis.5,6 The endocytotic vesicle with the 7-TMR can be targeted for lysosomal degradation or the receptor can be recycled back to the plasma membrane. β-Arrestin-dependent clathrin-mediated internalization of 7-TMRs appears to be a general mechanism of receptor desensitization for the majority of 7-TMRs. Depending upon the stability of interaction between β-arrestins and receptors, the receptors are distinguished as class A and class B receptors. Class A receptors, such as the β2-adrenergic receptor (β2-AR), transiently interact with β-arrestins and undergo rapid recycling. Class B receptors, such as the angiotensin receptor subtype 1 a (AT1aR), stably interact with β-arrestins and exhibit slow recycling. 7
β-Arrestins also serve as adaptor proteins for E3 ubiquitin ligase, which mediates ubiquitylation and degradation of 7-TMRs. 8 They interact with several E3 ubiquitin ligases, including Mdm2, neural precursor cell expressed developmentally down regulated 4 (NEDD4) and AIP4 to promote receptor ubiquitylation and degradation, such as β2-AR and chemokine receptor CXCR4.9–11 Recently, it has been determined that β-arrestins regulate ubiquitylation and internalization of certain non-7-TMR membrane proteins, such as TGF-β, insulin-like growth factor I receptor, calcium channels, the Na(+)/H(+) exchanger 1 and vascular endothelial cadherin.12–16
β-Arrestins function as signalosome adaptor/scaffolding proteins that regulate 7-TMR activation of MAPKs, including ERK1/2, 17 JNK, 18 p38 kinases, 19 and Src family kinases.20,21 In addition to MAPK regulation, β-arrestins also scaffold AKT, PI3 kinase and phosphodiesterase 4 upon activation of specific receptors.22–25 In contrast to the established function of β-arrestin as a G protein signaling terminator, 7-TMR can signal through β-arrestins without coupling to G proteins, a phenomenon referred to as ‘G protein-independent signaling’. β-Arrestin-mediated G protein-independent signaling exhibits distinct spatial temporal differences in the signaling cascade compared with G protein-dependent signaling. For example, β-arrestin-dependent activation of ERK 1/2 is more localized in the cytoplasm and exhibits a prolonged activation. In contrast, G protein-dependent transient activation of ERK 1/2 is distributed in both the cytoplasm and nucleus, and activates the transcription factor ELK-1. 26 β-Arrestin-dependent ERK activation activates cytoplasmic transcription factors signaling to the cell survival pathway.
The finding that β-arrestins can mediate G protein-independent signaling led to the discovery of ‘biased agonists’. Biased agonists can selectively activate G protein-dependent signaling or β-arrestin-dependent signaling. For example, stimulation of AT1aR by SII (Sar1, Ile4, Ile8-angiotensin II), a peptide analog of angiotensin II, led exclusively to β-arrestin-dependent signaling without activating G proteins.
27
Biased agonists can also activate G protein-dependent signaling. An example of the latter biased agonist is an agonist of the nicotinic acid receptor GPR109A, a 7-TMR which couples to Gi/Go proteins. Nicotinic acid activation of the GPR109A receptor has been used to treat dyslipidemia and appears to lower triglycerides via G protein-dependent signaling. However, a side effect of nicotinic acid is cutaneous flushing, which depends on β-arrestin 1.
28
Agonists of GPR109A, which selectively trigger G protein signaling without engaging β-arrestin 1, can maintain drug efficacy with fewer side effects. Taken together, the general functions of β-arrestins are summarized in Figure 1.
β-Arrestin 1 and 2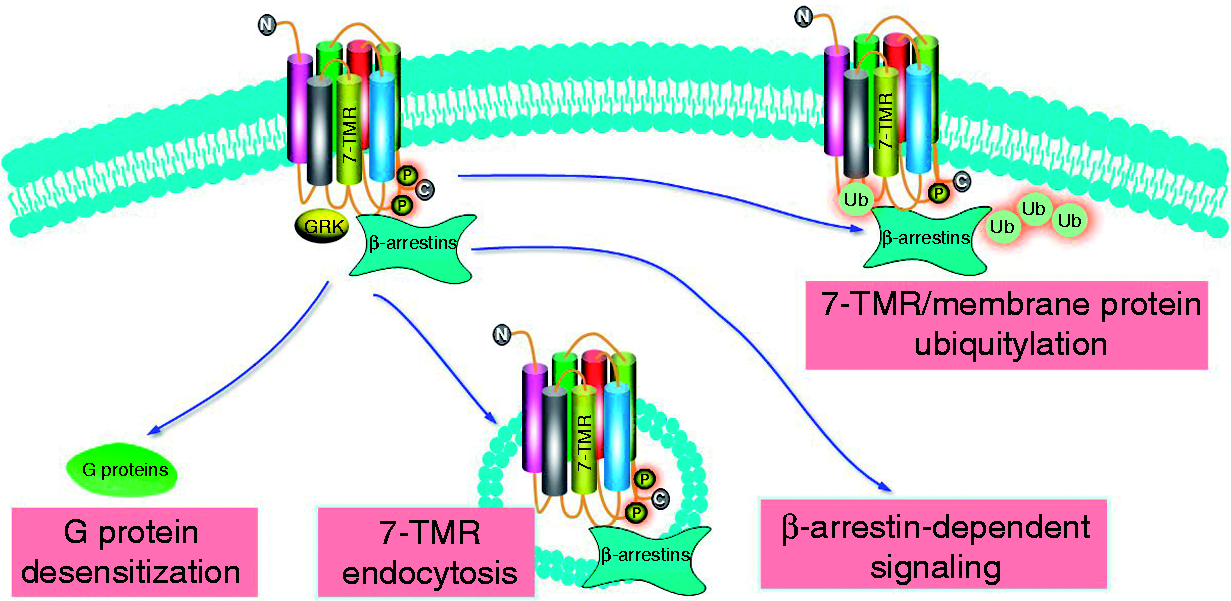
Isoform specific regulation of immune cells by β-arrestins
Macrophages and monocyte
β-Arrestin (β-arr) was altered in inflammation.
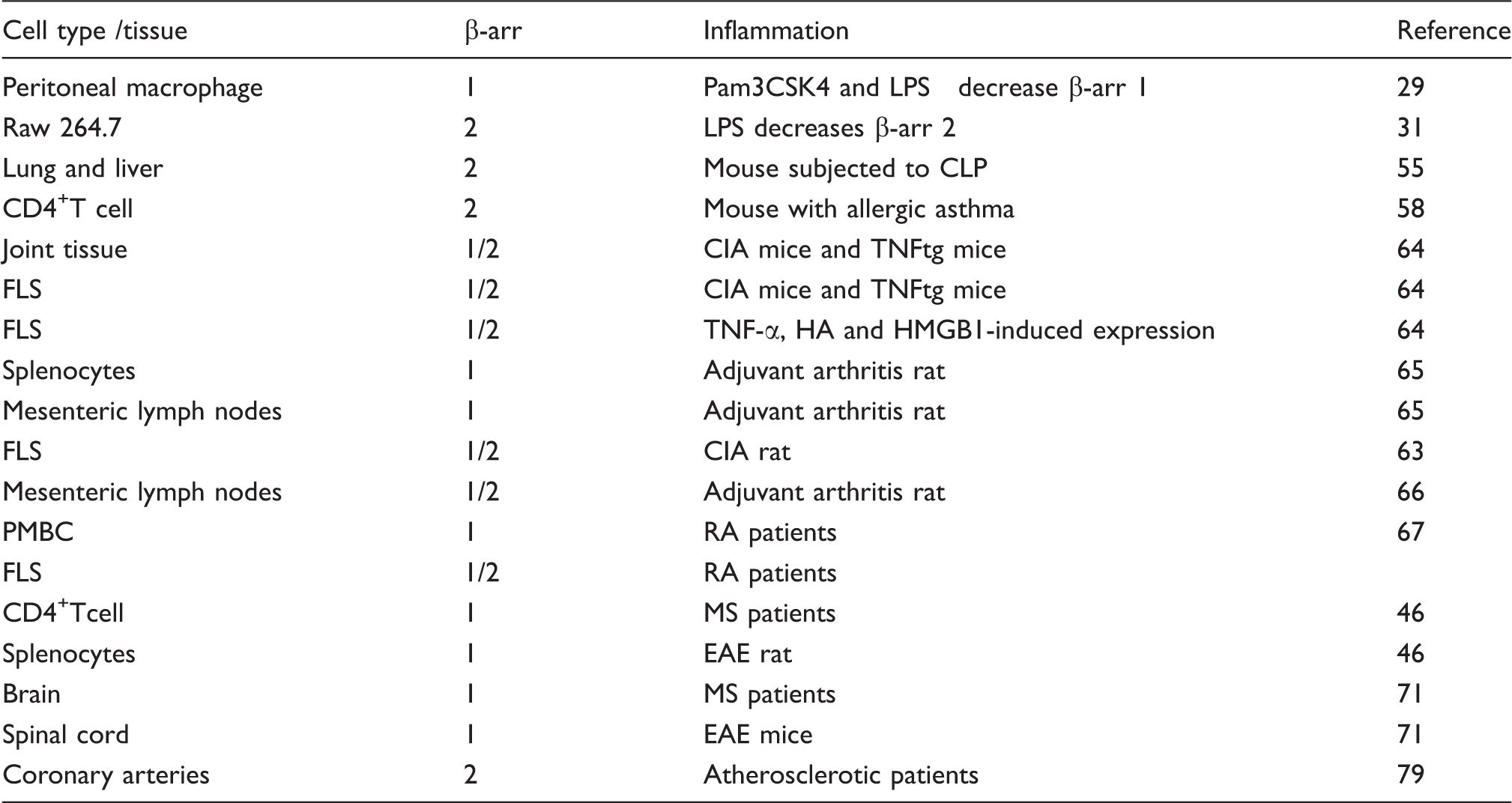
TLR-induced inflammation is regulated by β-arrestins. Wang et al. 30 reported that LPS-, polyI:C-, CpG DNA- and CD40L-induced TNF-α, IL-6 and IL-12p40 production were increased in bone marrow-derived macrophages isolated from β-arrestin 2 knockout (KO) mice. β-Arrestins directly interact and inhibit ubiquitination of TRAF6 following TLR or IL-1 receptor activation preventing TRAF6-mediated NF-κB activation. β-Arrestin 2 interacts with cytosolic IκBα and inhibits NF-κB activation and expression of its target gene, inducible NO synthase (iNOS), in response to LPS stimulation. These findings suggest that β-arrestin 2 stabilizes the NFκB/IκBα complex preventing NF-κB activation in macrophages. 31 Parameswaran et al. 32 demonstrated that in the Raw264.7 cells, knockdown of β-arrestin 1 leads to enhanced LPS-induced phosphorylation and degradation of NFκB1 p105, and enhanced MEK 1/2 and ERK 1/2 phosphorylation. Sender et al. 33 observed that surfactant protein-A enhances alveolar macrophage β-arrestin 2 expression, and inhibits LPS-induced TLR4 expression and co-localization of TLR4 with early endosome antigen 1 through β-arrestin 2. In this study, LPS-induced TNF-α release in bronchoalveolar lavage fluid was increased in β-arrestin 2 KO mice compared with WT mice. In splenocytes, β-arrestin isoform-specific regulation of cytokines. LPS-induced TNF-α, IL-6 and others were decreased in splenocytes from β-arrestin 1 KO mice compared with WT mice, while LPS-induced TNF-α and IL-6 were increased in splenocytes from β-arrestin 2 KO mice. 34 However, Porter et al. 35 showed that LPS-induced IL-1β, IL-12p70, IL-5 and INF-γ were decreased in CD11b+/CD11b− splenocytes from β-arrestin 2 KO mice compared with WT mice. In β-arrestin 1 KO mice, LPS-induced inflammatory gene expression was decreased in CD11b− splenocytes. 35 The disparate findings among these studies may be, in part, a result of different cells, for example, mixed cellular phenotypes in splenocytes versus CD11b+/CD11b− cells. β-Arrestins may regulate inflammatory responses in a cell phenotype and β-arrestin isoform-dependent manner.
Altered expression of β-arrestins (β-arr) regulates inflammation.
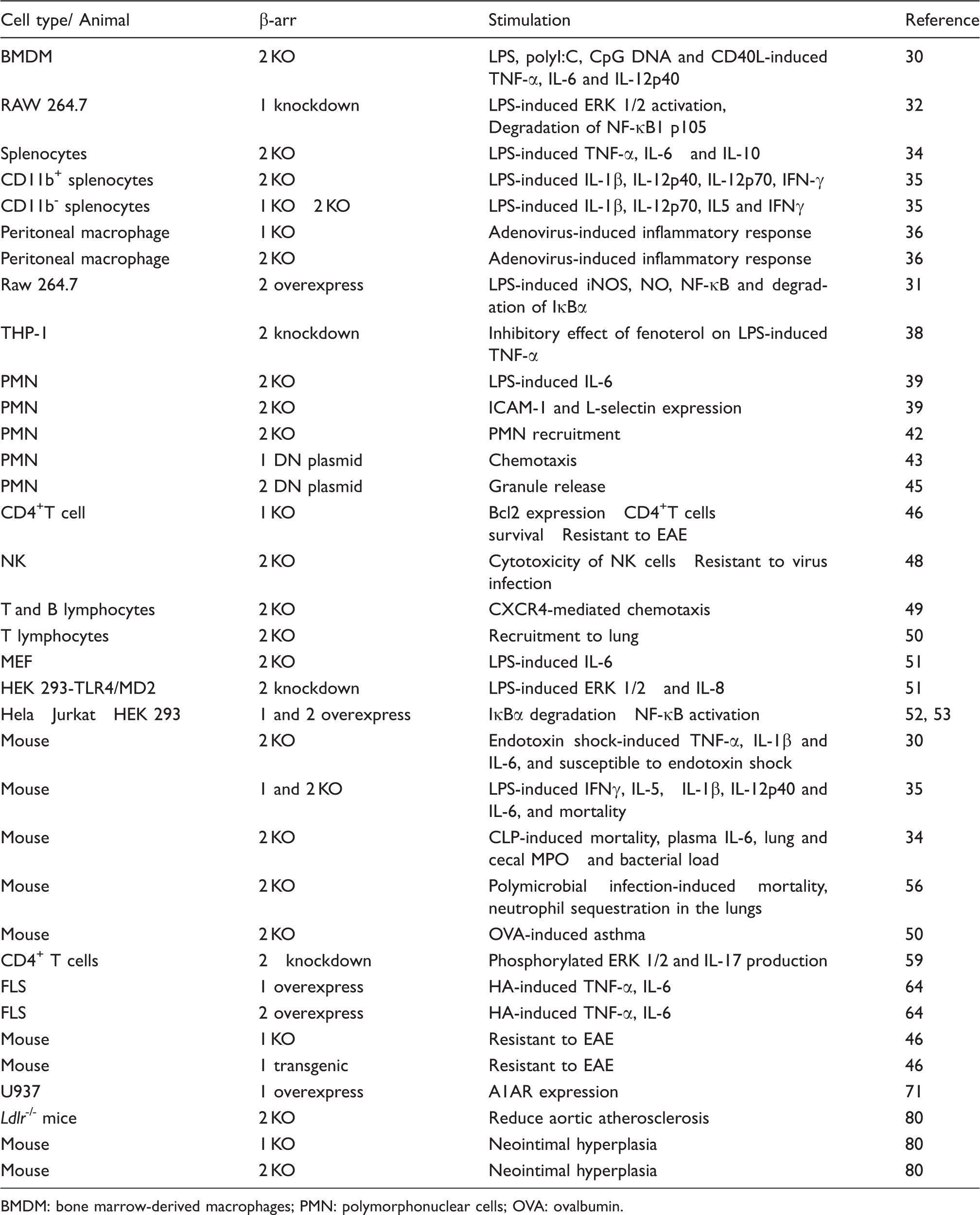
BMDM: bone marrow-derived macrophages; PMN: polymorphonuclear cells; OVA: ovalbumin.
Certain 7-TMRs can regulate TLR4 signaling through β-arrestins and vice versa. For example cross talk can occur between β2-AR and TLR signaling pathways in murine macrophage RAW 264 cells. 31 The expression of β-arrestin 2, which is required for β2-AR signaling, is reduced in RAW 264 cells after stimulation with LPS. 31 However, Fenoterol, a β2-AR agonist, inhibited LPS-induced TNF-α and IL-8, and decreased membrane TLR4/CD14 complex in THP-1 cells. The latter occurs through β-arrestin 2-mediated signaling as knockdown of β-arrestin 2 abrogated the suppression effect of fenoterol on LPS responses. 38 In general, the macrophage/monocytes studies reviewed suggest that β-arrestins 1 and 2 have distinct, and sometime opposing functions, in regulating inflammatory responses.
Neutrophils
Neutrophils are recruited to the sites of infection upon bacterial invasion and participate in the first line of defense through phagocytosis and bacterial killing. LPS-stimulated IL-6 production was increased in neutrophils from β-arrestin 2 KO mice compared with WT cells. β-Arrestin 2 deficiency resulted in an augmented expression of adhesion receptor ICAM-1 and
β-Arrestins are essential regulators of neutrophil chemotaxis. 40 Both β-arrestin 1 and 2 are required for IL-8-induced CXCR1 internalization, which, together with CXCR2, is responsible for IL-8-induced neutrophil chemotaxis. 41 In vivo studies in WT and β-arrestin 2 KO mice revealed that CXCR2-mediated neutrophil recruitment to sites of inflammation was increased in β-Arrestin 2 KO mice compared with WT mice. Su et al. 42 demonstrated that the deletion of β-arrestin 2 resulted in increased Ca2+ mobilization, superoxide anion production and GTPase activity in neutrophils, but decreased CXCR2 internalization relative to WT mice. CXCL1-induced recruitment of neutrophils was increased in β-arrestin 2 KO mice in two animal models: the dorsal air pouch model and the excisional wound healing model. 42 Ge et al. 43 demonstrated that neutrophil protease-activated receptor-2 (PAR-2) activation promotes ERK 1/2 and β-arrestin 1 dependent re-organization of the actin cytoskeletion, responsible for pseudopodia extension and chemotaxis. Prolonged ERK 1/2 activation in the pseudopodia is associated with a PAR-2/β-arrestin complex, which promotes chemotaxis. 43 Therefore, β-arrestins may regulate chemotaxis in a cell phenotype and receptor-dependent manner. For a more in depth review of roles of β-arrestins in neutrophils, lymphocytes, and macrophage chemotaxis and migration, the reader is referred to comprehensive reviews by Luttrell and Gesty-Palmer., 26 DeFea 40 and DeWire et al. 44
β-Arrestins not only regulate chemotaxis to specific chemokines, but are also essential for chemokine-induced granule exocytosis, which is important for neutrophil function. Chemoattractant-stimulated granule release from neutrophils, basophils and eosinophils is critical for innate immune responses. In addition to chemotaxis, β-arrestins 1 and 2 regulate neutrophil degranulation. 45 Barlic et al. 45 demonstrated that IL-8 stimulates rapid formation of β-arrestin complexes with tyrosine kinase Hck or cFgr. Formation of β-arrestin-Hck complexes leads to Hck activation and trafficking of the complexes to granule-rich regions. Granulocytes expressing a dominant-negative β-arrestin mutant did not release granules or activate tyrosine kinases after IL-8 stimulation. 45
Lymphocytes
CD4+ T cells are essential for adaptive immunity, and dysregulation of CD4+ T cell has been implicated in autoimmunity. Shi et al. 46 demonstrated that β-arrestin 1 promotes CD4+ T cell survival. Enhanced cell survival was thought to be a consequence of β-arrestin 1-dependent expression of proto-oncogene Bcl2.
NK cells are also crucial in the innate immune responses through surveillance of transformed and infected cells. 47 NK cells contain abundant granules with cytolytic proteins, which induce the death of target cells. 47 Yu et al. 48 demonstrated that β-arrestin 2 associates with KIR2DL1, an inhibitory receptor of NK cells. The receptor recognizes MHC class I on target cells, which confer protection to healthy cells. Upon ligand activation, β-arrestin 2 mediates recruitment of the tyrosine phosphatases SHP-1 and SHP-2 to KIR2DL1, and facilitates inhibitory signaling. β-Arrestin 2 KO mice exhibit increased cytotoxicity of NK cells, and these mice are less susceptible to murine cytomegalovirus infection. The latter effects were abolished by depletion of NK cells compared with WT mice. These composite findings suggest that β-arrestin 2 inhibits activation of NK cells. 48
β-Arrestin 2 positively regulates T lymphocyte trafficking: using β-arrestin 2 deficient mice, Fong et al. 49 demonstrated that β-arrestins are required for the chemokine receptor CXCR4-mediated CD3+ T and B lymphocyte chemotaxis in vitro. Another in vivo study demonstrated that CD4+ T cell recruitment to the lung was impaired in β-arrestin 2 KO mice with allergic asthma. 50
Other cells
Mouse embryonic fibroblasts (MEF) from WT, β-arrestins KO, β-arrestins 1 and 2 double KO, and MEFs with reconstituted WT β-arrestins in the double KO cells have been studied. β-Arrestin 2 positively regulates LPS-induced IL-6 production. 51 The effect of small interfering RNA (siRNA)-specific knockdown of β-arrestins or overexpression of WT β-arrestins in HEK 293 cells co-transfected with TLR4 and MD2 was also investigated. These studies demonstrated that β-arrestin 2 positively regulates LPS-induced ERK 1/2 activation and IL-8 production. However, both β-arrestins 1 and 2 negatively regulated LPS-induced NF-κB activation. 51 The multiple effects of β-arrestins on ERK 1/2 signaling pathway activation in cells exceeds the scope of the present review and has been comprehensively reviewed by Luttrell and Gesty-Palmer 26 and DeWire et al. 44 The inhibitory effect of β-arrestins 1 and 2 on NF-κB activation is consistent with other studies. Witherow et al. 52 demonstrated that both β-arrestins 1 and 2 interact with IκBα and inhibit NF-κB activation in Hela cells and the T lymphocyte cell line Jurkat cells. Over-expression of β-arrestins 1 and 2 attenuated activation of NF-κB. 52 Gao et al. 53 also reported that β-arrestin 2 interacting with IκBα prevents the phosphorylation and degradation of IκBα and inhibits NF-κB activation in HEK 293 cells. Additionally, stimulation of β2-AR enhances β-arrestin 2 IκBα interaction and promotes stabilization of IκBα. 53 The divergent effect of β-arrestins on ERK activation versus NF-κB signaling in the context of gene expression is uncertain. However, considering the scope of inflammatory genes regulated by NF-κB, β-arrestin inhibition of this signaling pathway may be predominately anti-inflammatory.
β-Arrestins regulate inflammatory disease
Endotoxemia and sepsis
Studies have implicated β-Arrestins in TLR signaling and gene activation. β-Arrestins 1 and 2 interact directly with TRAF6, a critical mediator in TLR signaling, preventing auto-ubiquitination of TRAF6 and activation of NF-κB and AP-1.
30
Compared with WT mice, β-arrestin 2 KO mice subjected to endotoxin shock exhibit higher expression of pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6. β-Arrestin 2 KO mice sensitized to LPS by
In contrast to LPS shock, polymicrobial sepsis induced by cecal ligation and puncture (CLP) is generally accepted as a clinical relevant animal model. 54 Fan et al. 34 demonstrated that β-arrestin 2 KO mice were more susceptible to CLP-induced sepsis. When β-arrestin 2 KO and WT mice were subjected to CLP, survival rate was significantly decreased in β-arrestin 2 KO mice compared with WT mice. Subsequent studies demonstrated that CLP-induced plasma IL-6 and pulmonary and cecal myeloperoxidase (MPO) activity were significantly increased in the β-arrestin 2 KO mice. Also bacterial load in peripheral blood, peritoneal fluid and lung tissue were also significantly increased in the mice. 34 Recent observations in another CLP sepsis study demonstrated that β-arrestin 2 levels were decreased in the lung and liver post-CLP. Flavocoxid, a dual inhibitor of COX-2 and 5-LOX, preserved β-arrestin 2 expression and decreased NF-κB activation. 55 Another recent article supports the notion that β-arrestin 2 negatively regulates systemic inflammation in response to polymicrobial infection. β-Arrestin 2 KO mice exhibit increased neutrophil sequestration and inflammation in the lungs following polymicrobial infection. 56 Thus, β-arrestin 2 appears to be a negative regulator of inflammation in polymicrobial sepsis.
Asthma
Allergic asthma is a chronic inflammatory disorder of the airways that is characterized by migration of T helper cells and eosinophils into the lung. Migration of Th2 cells and eosinophils to the lung is important to their inflammatory function and is regulated, in large part, by chemokine receptors. Compared with WT mice, allergen-sensitized β-arrestin 2 KO mice do not accumulate CD3+ T lymphocytes in their airways, nor do they demonstrate other inflammatory features characteristic of asthma. In contrast, the airway inflammatory response to LPS, an event not coordinated by Th2 cells, was unaffected in β-arrestin 2 KO mice. In another asthma study β-arrestin 2 KO mice demonstrated defective chemokine-mediated CD4+ T cell migration to the lung. 50 In subsequent studies the authors demonstrated that β-arrestin 2 also mediates the migration of hematopoietic-derived eosinophils to the lung. 57 Additionally, it has been demonstrated that CD4+ T lymphocytes of mice with allergic asthma express higher levels of β-arrestin 2 mRNA and protein. 58 Treatment of CD4+ T lymphocytes with siRNA targeting the β-arrestin 2 down-regulated phosphorylated ERK1/2 and IL-17 production essential to the pathogenesis of asthma. 59 A recent study by Nichols et al. 60 clarified the role of β-arrestin 2 in proteinase-activated receptor-2 (PAR2) signaling in the ovalbumin-induced murine model of allergic asthma. PAR2 is reported to have both protective and pro-inflammatory effects in the airway. The pro-inflammatory responses are mediated by β-arrestin 2, whereas the protective effects are not. 60 As β-arrestin 2 appears to augment allergic inflammation, therapies focused on β-arrestin 2 may prove useful in the treatment of asthma.
Chronic use of β-agonists by asthmatics is associated with a loss of bronchodilator effects. β-Arrestin 2 KO mice exhibit augmented β-agonist-mediated airway smooth muscle relaxation and β-agonist-stimulated cAMP production. 61 Moore et al. 62 demonstrated that Salmeterol, a long-acting β2-AR agonist, did not induce significant β2-AR internalization or degradation, and was incapable of stimulating the translocation of β-arrestin 2 to the cell surface. Thus, the differing efficacy of β-agonists in asthma may be a consequence of differences in activation of β-arrestin-dependent signaling. These studies suggest a role of the β2-AR/ β-arrestin 2 axis in regulation of bronchial smooth muscle relaxation in experimental models of asthma.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic autoimmune joint disease involving activation of innate immunity and dysfunction of adaptive immunity. A study by Wang et al. 63 demonstrated that both β-arrestin 1 and 2 levels were increased in fibroblast-like synoviocytes (FLS) from collagen-induced arthritis (CIA) rats. Using a CIA mouse model and a human TNF-α transgenic (TNFtg) mouse model, Li et al. 64 demonstrated that β-arrestin 1 and 2 expression was significantly increased in joint tissue of both CIA and TNFtg mice. 64 The protein and mRNA levels of β-Arrestin 1 and 2 were also increased in FLS isolated from knee joints of CIA mice. TNF-α and low molecular mass hyaluronan (HA) induced an increase of β-arrestin 1 and 2 expression in FLS, while high mobility group box (HMGB)-1 selectively stimulated β-arrestin 1 expression. In FLS, HA-induced TNF-α and IL-6 production were increased by over-expression of β-arrestin 1, but decreased by overexpression of β-arrestin 2. Thus, β-arrestin expression is altered in FLS during inflammation and β-arrestins isoform specifically regulate FLS inflammatory responses. 64 To examine the in vivo role of β-arrestin 2 in pathogenesis of arthritis, WT and β-arrestin 2 KO mice were subjected to collagen antibody-induced arthritis (CAIA). β-Arrestin 2 KO mice exhibited more severe arthritis in CAIA. Therefore, β-arrestin 2 may be anti-inflammatory in CAIA. 64
Splenocytes from adjuvant arthritis rats on d 18 after induction of the disease exhibit increased β-arrestin 1 expression compared with controls. The level of β-arrestin 1 in splenocytes returned to normal at d 45. A similar time course for β-arrestin 1 expression was observed in mesenteric lymph nodes. 65 In another study, the total glucosides of paeony (TGP), a herbal drug, decreased hind paw swelling and arthritis severity of CIA rats, and significantly reduced the expression of β-arrestins. 63 In a subsequent study, it was demonstrated that β-arrestin 1 and 2 levels were increased in mesenteric lymph nodes from adjuvant arthritis rats. 66 Although the data obtained with TGP are only correlative with reduction in both β-arrestin isoforms, the combined results with other studies with in vitro overexpression studies and β-arrestin 2 KO suggest that β-arrestin 1 may be pro-inflammatory versus β-arrestin 2, which maybe anti-inflammatory in experimental arthritis.
The relevance of all these experimental findings in animal models to human RA remains to be determined. However, β-arrestin 1 and 2 expression in tissues from RA patients has been examined. Recent studies demonstrate that both β-arrestin 1 and 2 expression is increased in FLS from RA patients compared with healthy controls (unpublished data). However, Lombardi et al. 67 observed that β-arrestin 1 expression exhibited no difference in PBMC from RA patients compared with healthy donors. It is possible that in clinical RA localized changes in joint tissue β-arrestin expression may be more predictive of pathologic outcome.
Multiple sclerosis
Multiple sclerosis (MS) is characterized by the presence of plaques of demyelination throughout the central nervous system. Although the etiology of the disease has not been established, it is believed to involve autoimmune mechanisms. Ohguro et al. 68 discovered that sera from MS patients formed an autoimmune complex with β-arrestin 1, while such serum auto-Abs were not detected from patients with other neurological diseases. The authors suggested that serum β-arrestin 1 Ab titers may be useful for the diagnosis and evaluation of the disease’s course.68,69 Forooghian et al. 70 demonstrated that MS patients had a greater prevalence of positive T cell proliferative responses to β-arrestin 1 than healthy controls. Thus, β-arrestin has been identified as a novel non-myelin auto-antigen in MS. Such a notion is supported by an animal model of MS. In an experimental autoimmune encephalomyelitis (EAE) rat model, it has been demonstrated that β-arrestin 1 levels were significantly increased in the splenocytes from EAE compared with controls. β-Arrestin 1 positively regulates naïve and activated CD4+ T cell survival through regulation of acetylation of histone H4 at the Bcl2 promoter. β-Arrestin 1 KO mice exhibit significantly decreased numbers of CD4+ T cells in the spleen and lymph nodes. β-Arrestin 1 KO mice were much more resistant to EAE, whereas overexpression of β-arrestin 1 increased susceptibility to this autoimmunity. 46 The authors also observed that CD4+ T cells from patients with MS had much higher expression of β-arrestin 1, whereas knockdown of β-arrestin 1 in those cells increased apoptosis. Therefore, the authors concluded that β-arrestin 1 is a critical for CD4+ T cell survival and is a factor in susceptibility to autoimmunity. 46
In another clinical study, Tsutsui et al. 71 further demonstrated that β-arrestin 1 expression was increased in brains of MS patients and varied inversely with the A1 adenosine receptor (A1AR). In vitro studies demonstrated that β-arrestin 1 overexpression down-regulated A1AR expression. 71 In murine EAE, β-arrestin 1 and A1AR expression in the spinal cord displayed a similar inverse pattern compared with that observed in MS brain. EAE-induced neuroinflammation and neurobehavioral deficits were suppressed by glucocorticoid treatment, and correlated with concurrent reductions in β-arrestin 1, but enhanced A1AR expression. These studies suggest that β-arrestin 1 and A1AR expression and function are critical determinants in MS neuroinflammation. 71
Studies have shown that β-arrestin 1 may down-regulate other receptors relevant to MS pathogenesis. β2-AR regulates astrocyte glucose metabolism to maintain brain activity in both normal and stress conditions. 72 Clinical studies demonstrated that β2-AR is decreased in plaques and white matter of MS patients in postmortem brain sections compared with non-neurologic disease patients.73,74 As β-arrestin 1 regulates β2-AR internalization and degradation, increased β-arrestin 1 expression may result in decreases of β2-AR and reducing its neuroprotective effect.31,75
Inflammatory bowel disease
Inflammatory bowel disease includes both Crohn’s disease and ulcerative colitis, which are characterized by chronic relapsing inflammation of the gastrointestinal tract. 76 Lee et al. 77 demonstrated that β-arrestin 1 KO mice exhibit attenuated disease pathology in the experimental ulcerative colitis induced by trinitrobenzene sulfonic acid (TNBS) or dextran sulfate sodium. 77 IL-6 expression in the plasma and colon tissue were also decreased in β-arrestin 1 KO mice compared with WT mice subjected to experimental colitis. These data suggest that β-arrestin 1 mediates IL-6 expression in the colon in experimental colitis model. 77 In addition to β-arrestin 1, Fan et al. 78 demonstrated that in the experimental ulcerative colitis in rats induced by TNBS, β-arrestin 2 expression was significantly decreased in the splenic lymphocytes. As β-arrestin 2 is frequently anti-inflammatory in a variety of diseases, these data suggest that β-arrestin 2 may also play a role in ulcerative colitis development.
Atherosclerosis and neointimal hyperplasia
Atherosclerosis and associated cardiovascular diseases are among the leading causes of mortality and morbidity worldwide. Cardiovascular risk factors (e.g. dyslipidemia, hypertension, diabetes and smoking) initiate chronic inflammatory responses that cause endothelial dysfunction, a key process in the initiation and progression of atherosclerosis. Transcriptional profiling data in severely atherosclerotic and non-atherosclerotic human coronary arteries showed that β-arrestin 2 mRNA levels are twofold higher in atherosclerotic patients than non-atherosclerotic controls. 79 Atherosclerosis and arterial injury-induced neointimal hyperplasia involve smooth muscle cell (SMC) proliferation and migration into the arterial intima. ldlr−/− mice have an elevated serum cholesterol level and are employed as an animal model of atherosclerosis. Kim et al. 80 demonstrated that deficiency of β-arrestin2 in ldlr−/− mice reduced aortic atherosclerosis and decreased the prevalence of atheroma SMCs.
Carotid endothelial denudation is employed to induce neointimal hyperplasia and has been studied in congenic WT, and β-arrestin 1 2 KO mice. 80 Neointimal hyperplasia was enhanced in β-arrestin 1 KO mice, but diminished in β-arrestin 2 KO mice. After carotid injury, SMC ERK 1/2 activation and proliferation were also increased in β-arrestin 1 KO and decreased in β-arrestin 2 KO mice. Concordantly, SMC thymidine incorporation, ERK 1/2 activation and SMC migration evoked by 7-TMRs were greater in β-arrestin 1 KO than WT, but less in β-arrestin 2 KO. These studies support the hypothesis that SMC proliferation and migration in atherosclerosis are regulated reciprocally by β-arrestin 2 and β-arrestin 1.
Conclusions
The expression of β-arrestins 1 and 2 is regulated in immune cells in response to diverse inflammatory conditions. In turn, β-arrestin 1 and 2 regulate diverse inflammatory responses and contribute to multiple inflammatory diseases. Further evaluation of the effect of β-arrestins 1 and 2 in different cells at different stages of inflammatory diseases may generate a clearer picture how these unique proteins change in inflammation. The function of β-arrestins 1 and 2 in regulation of inflammation can be quite different. β-Arrestin isoforms regulate distinct spatial and temporal activation of signaling proteins that activate different target genes. β-Arrestin 1 or 2 KO mice have provided an important investigative approach and have been employed extensively. However, it is also possible that the β-arrestin isoforms can compensate for each other as knockout of single β-arrestin 1 or 2 in mice appears not to affect the mice, while knockout of both β-arrestin 1 and 2 is lethal. The availability of conditional β-arrestin 1 and 2 KO mice could help resolve this problem. Differentiation of signaling mediated by β-arrestins 1 and 2 in different relevant immune cells remains a huge challenge. Additionally, how β-arrestin levels are regulated at the transcriptional and/or translational level in inflammation remains poorly understood. Further investigations of the role of β-arrestins in inflammation are warranted and may lead to novel approaches to treat a variety of inflammatory diseases.
Footnotes
Acknowledgements
Thanks to Drs James Cook and Perry Halushka for their helpful suggestions.
Funding
This work was supported, in part, by GM27673 (JAC) and AI079248 (HF).