
Abstract
Microparticles (MPs) are small, membrane-bound vesicles that arise from dead and dying cells, and display pro-inflammatory and pro-thrombotic activity. As shown previously, the RAW 264.7 murine macrophage cell line can release MPs following stimulation with LPS or polyinosinic:polycytidylic acid [poly (I:C)], ligands of TLR4 and TLR3 respectively. To determine the relationship of these MPs to those released during apoptosis, the nucleic acid content of MPs from cultures stimulated with LPS or poly (I:C) was compared with the nucleic acid content of MPs from untreated cells or cells induced to undergo apoptosis by treatment with etoposide or staurosporine (STS). As results of these studies showed, MPs from activated, apoptotic and untreated cells had features in common, as demonstrated by binding of the nucleic acid dyes SYTO 13 and propidium iodide; molecular mass of DNA; and binding of monoclonal anti-DNA and anti-nucleosome Abs. While MPs from the different culture conditions all contained ribosomal RNA, ribosomal RNA from MPs from STS-treated cells showed cleavage and degradation. Taken together, these studies indicate that the nucleic acid content of MPs from activated and apoptotic cells have important similarities, suggesting that events during TLR activation may lead to apoptosis and subsequent MP release.
Introduction
Microparticles (MPs) are small, membrane-bound vesicles that arise from activated and dying cells by a blebbing process.1–3 These particles range in size from 0.1 to 1.0 microns, and contain nuclear, cytoplasmic and membrane components. As shown in in vitro and in vivo experiments, MPs have diverse biological functions and can mediate inflammation, thrombosis and information exchange between cells, among other activities. As such, MPs may play an important role in physiological and pathophysiological settings.4–8 Furthermore, MPs can serve as biomarkers of disease pathogenesis, with their levels in blood reflecting events in a wide range of tissue. 9
While the function of blebbing is not fully known, this process may regulate the surface-to-volume ratio of cells as they shrink during death. Like the formation of apoptotic bodies, blebbing may facilitate the clearance of material from dead and dying cells.10–12 Nevertheless, despite the close link between blebbing and apoptosis, cell activation can also lead to particle generation. For immune and other hematopoietic cells, however, the distinction between activation and death can be difficult because of the occurrence of activation-induced cell death. Among triggers for activation, LPS and other ligands of the TLRs can induce apoptosis or pyroptosis of macrophages among other cell types, with cell death a likely mechanism of host defense to limit intracellular infection.13–15
In a previous study, we assessed the mechanisms of MP release during the activation of macrophages by TLR ligands. 16 These studies demonstrated that both LPS and polyinosinic:polycytidylic acid [poly (I:C)] can induce MP production, as assessed by flow cytometry. This release depended on NO and could be blocked by inhibitors of inducible nitric oxide synthase (iNOS). The MPs arising during TLR stimulation bound annexin V and, by flow cytometry, showed a size distribution consistent with particles from other sources.
While TLR ligands can potently induce macrophage activation, previous studies with RAW 264.7 cells indicated a significant increase in apoptosis in the stimulated cultures. 16 Furthermore, inhibition of caspases by the caspase inhibitor Z-VAD-FMK caused an increase in the number of MPs from RAW 264.7 cells undergoing activation. This agent can cause a paradoxical increase in cell death because of its effects on the NUR77 orphan receptor, which undergoes cleavage by a caspase-like enzyme. With this enzyme blocked, necrosis ensues.17,18 These results support a close link between activation induced by TLR stimulation and cell death, and the role of regulatory systems in determining this balance.
Among their constituents, MPs contain nucleic acids, both DNA and RNA, as demonstrated by biochemical assays, as well as staining of particles with antinuclear Abs.19,20 While translocation of DNA into blebs is the likely source of this molecule in MPs from apoptotic cells, RNA may arise from the cytoplasm as the bleb forms. In contrast, the source of DNA in MPs arising during cell activation is less clear unless stimulation of TLRs or other receptors caused cell death; in this circumstance, the generation of blebs would resemble that occurring during classical apoptosis. Elucidating the nature of this DNA is important in determining mechanisms for MP release and the generation of extracellular DNA and its functional and antigenic properties.
In the current experiments, we have characterized the properties of MPs released by RAW 264.7 cells undergoing activation by LPS or poly (I:C), or apoptosis with etoposide or staurosporine (STS). We have analyzed the DNA and RNA content using biochemical assays, and assessed the antigenicity of the DNA and associated nucleosomal molecules by flow cytometry. As the results of these studies show, MPs from both activated and apoptotic cells contain DNA and RNA, with the DNA accessible to binding by Abs. These results suggest that particle release during macrophage activation may result from cell death, emphasizing the link between activation and cell death, and the role of TLR stimulation in inducing the production of extracellular DNA.
Materials and methods
Cell culture
RAW 264.7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and were maintained in RPMI 1640 medium (Invitrogen Life Technologies, New York, NY, USA) supplemented with 10% FBS (Atlanta Biologicals, Lawrenceville, GA, USA) and 20 µg/ml gentamicin (Invitrogen Life Technologies) at 37℃, 5% CO2. Approximately every 2–3 d, cells were sub-cultured upon reaching 80% confluence.
Analysis of microparticles by flow cytometry
Briefly, RAW 264.7 cells from a culture at 80% confluence were plated in RPMI 1640 medium supplemented with 10% FBS and 20 µg/ml gentamicin (complete media) at a density of 2.2 × 106/well of a six-well plate. Cells were incubated for 20 h, after which they were washed twice with 1 × PBS (calcium and magnesium-free; Invitrogen Life Technologies) and incubated in phenol red-free Optimem (Invitrogen Life Technologies). At this point, agents to induce activation or apoptosis were added, yielding final concentrations of 30 µg/ml etoposide, 50 µg/ml LPS, 1 μM STS (all purchased from Sigma-Aldrich, St. Louis, MO, USA) or 250 ng/ml high molecular mass poly (I:C) (Invivogen, San Diego, CA, USA). After 20–22 h, culture supernatants were removed by aspiration and centrifuged at 500 g for 5 min. The resulting supernatants were immediately assayed for MP quantity and phenotype. Alternatively, samples for lactate dehydrogenase (LDH) measurement and DNA content were stored at –20℃ until assayed.
Samples were analyzed on the side scatter detection setting using a FACScan flow cytometer (Becton Dickinson, Mansfield, MA, USA). The acquisition software used was FlowJo Collectors Edition (Tree Star, Ashland, OR, USA). The samples were diluted with 1 × PBS to produce solutions in which events were detected at a maximum rate of 1000 MPs per second. The dilutions ranged from 1 : 5 to 1 : 50, depending on MP concentration. By using pre-determined parameters, events above a minimum size threshold were collected. Data were collected until 10,000 events were counted or 1 min of counting had elapsed. Background events for buffer alone were subtracted and the counts/μl of sample was calculated using the recorded flow rate of the machine.
Samples were stained separately with SYTO 13 green fluorescent nucleic acid stain (Invitrogen Life Technologies), PI (Sigma-Aldrich) or FITC-labeled annexin V (BD Biosciences, Franklin Lakes, NJ, USA). For each staining condition, 300 μl was prepared; all samples were diluted as described above, with samples stained with SYTO 13 or propidium iodide (PI) diluted in 1 × PBS (calcium- and magnesium-free), and samples stained with FITC-labeled annexin V were diluted in annexin binding buffer (140 mM NaCl, 10 mM Hepes and 2.5 mM CaCl2). For 300 μl of diluted sample, the following volumes of staining reagent were added: 6 µl of 5 µM SYTO 13, 3 µl of 1 mg/ml PI and 3 µl of FITC-annexin V Ab solution. The data were analyzed using FlowJo analysis software.
Measurement of LDH activity
The activity of LDH in culture supernatants was determined using the Cytotox96 non-radioactive cytotoxicity assay (Promega, Madison, WI, USA). Supernatants were diluted 1 : 10 in 1 × PBS (calcium- and magnesium-free), and the assay was completed per kit instructions.
Quantitation of DNA release
The amount of double-stranded DNA released from activated or dying RAW 264.7 cells was measured using the Quant-iT PicoGreen dsDNA Reagent (PicoGreen; Invitrogen Life Technologies). Briefly, the supernatants were diluted 1 : 5 with 1 × TE [10 mM Tris-HCl and 1 mM EDTA (pH 8.0)]. Samples were incubated at a 1 : 1 ratio with the PicoGreen reagent diluted 1 : 200 in 1 × TE in black 96-well plates (Greiner Bio-One, Kremsmunster, Austria) for 5 min. Fluorescence was measured using a Tecan GENios microplate reader (Salzburg, Austria), with an excitation wavelength of 485 nm and an emission wavelength of 535 nm. Values were obtained as relative fluorescence units (RFU). The concentration of DNA (ng/ml) was calculated according to fluorescence values for a standard solution of double-stranded DNA from calf thymus (Worthington Biochemical Corporation, Lakewood, NJ, USA).
Purification of RNA and DNA from MPs
Briefly, actively growing RAW 264.7 cells were plated at a density of 2–3 × 106 cells per T175 tissue culture flask in complete medium and incubated overnight (20–22 h) at 37℃, 5% CO2. Between one and three flasks per treatment were used depending on expected yields of MPs. After 18 h culture monolayers were rinsed twice with 1 × PBS and then incubated in 25 ml/flask of phenol red-free Optimem (Invitrogen Life Technologies). Cells were then treated to induce MP production [50 µg/ml LPS, 250 ng/ml poly (I:C), 30 µg/ml etoposide or 1 μM STS]. The treated cells were incubated for 20–22 h at 37℃, 5% CO2. The medium was then removed from the cells and centrifuged twice at room temperature for 5 min at 500 g to remove any contaminating cells.
The cell-free supernatant was then passed through a 1.2 µm syringe filter (Pall Corporation, Cornwall, UK) to ensure removal of large contaminating material. MPs were then isolated from the filtered supernatant by ultracentrifugation at 10℃, 150,000 g for 25 min. The MP-containing pellets were re-suspended in cold 1 × PBS and ultra-centrifuged under the same conditions. Supernatants were carefully removed and the purified MPs were stored at –80℃. RNA and DNA were isolated from separate preparations of MPs. Following the thawing of the MP pellets, RNA and DNA were isolated using RNeasy mini kits (Qiagen, Valencia, CA, USA) and Apoptotic DNA Ladder kits (Roche, Indianapolis, IN, USA) respectively. The minimum recommended elution volumes were used to keep recovered RNA and apoptotic DNA concentrated for subsequent applications.
Nucleic acid purification and analysis
Isolated RNA was quantitated using Quant-iT Ribogreen RNA Reagent (Ribogreen reagent). Briefly, samples were prepared at a range of dilutions (1 × TE). They were then incubated at a 1 : 1 ratio with a 1 : 200 dilution of Ribogreen reagent in a black 96-well plate. The incubation took place in the absence of light for 5 min. The fluorescence produced was measured using a fluorescence plate reader (excitation 485 nm, emission 535 nm). Values were obtained as RFU. The RNA concentrations (ng/ml) were calculated according to fluorescence values for standard solutions made from ribosomal RNA (Roche). The samples were diluted to 2 ng/ml with DNAse- and RNAse-free water. The samples were submitted to the Duke University DNA Microarray Core Facility for analysis of RNA integrity using an Agilent Bioanalyzer (Picochip) using eukaryote total RNA detection settings.
DNA isolated from MPs was quantitated using PicoGreen reagent as described above. DNA electrophoresis was performed using a 1% agarose gel in 1 × TRIS Acetate-EDTA (TAE), (0.04M TRIS acetate, 0.001m EDTA) stained with ethidium bromide. Equal amounts of DNA were loaded per lane. Images of gels stained with ethidium bromide were photographed with a digital imaging system from Alpha Innotech (San Leandro, CA, USA).
MP binding of anti-DNA and anti-histone Abs
MPs released from activated or dying RAW 264.7 cells were purified using the same method described above, although cell numbers and culture size were reduced because the number of MPs required was much lower than those needed for RNA and DNA extraction experiments. After purification, MPs were re-suspended in 200 μl 1 × PBS (calcium- and magnesium-free). MP numbers were then determined by flow cytometry (as described above). Potential Fc receptor sites on the MPs were blocked by incubating the MPs with a rat anti-mouse CD16/CD32 (BD Bioscience) at a concentration of 0.5 µg/300,000 MPs for 15 min on ice. MPs were aliquoted into FACS tubes at a concentration of 50,000 per tube. The MPs were stained with monoclonal Abs that recognize DNA (QB1 and PL9-11), nucleosomes (PR1-3 and PA1) or isotype controls for 1 h at room temperature, followed by a F(ab')2 sheep anti-mouse IgG PE (Sigma) (1 h, room temperature, in the dark). The samples were then analyzed by flow cytometry.
Statistical analysis
A two-tailed, Student’s t-test analysis was used to analyze data between different treatment conditions. A value of P < 0.05 was considered significant.
Results
As a model for generating MPs by macrophages, RAW 264.7 cells were used, treating cells with LPS or poly (I:C) to activate TLR4 and TLR3, respectively, or with etoposide or STS to induce apoptosis under conditions previously used to assess MP generation by these cells.16,19 In these experiments, we stimulated cultures with LPS at a concentration of 50 µg/ml based on previous studies’ experiments, which showed that MP production stimulation by LPS was dose-dependent, with a concentration of 50 µg/ml leading to the highest number of particles.
16
Following incubation with the various stimulants, culture supernatants were obtained and analyzed for MPs. LDH and DNA were assessed as a measure of cell death. The number of MPs was determined by flow cytometry. As shown in Figure 1, irrespective of source, particles showed a similar pattern when assessed by forward scatter and side scatter.
Measurement of MPs by flow cytometry. RAW 264.7 cells were treated to induce activation [LPS or poly (I:C)] or apoptosis (etoposide or STS) for 20–22 h. After centrifugation to remove cells, the number of MPs was detected by flow cytometry. The dot blot indicates the profile of particles by forward and side scatter. In these experiments, calibration using sizing beads allowed the determination of flow cytometer parameters to allow detection of events only found within the size range (0.2–1 µm) of MPs.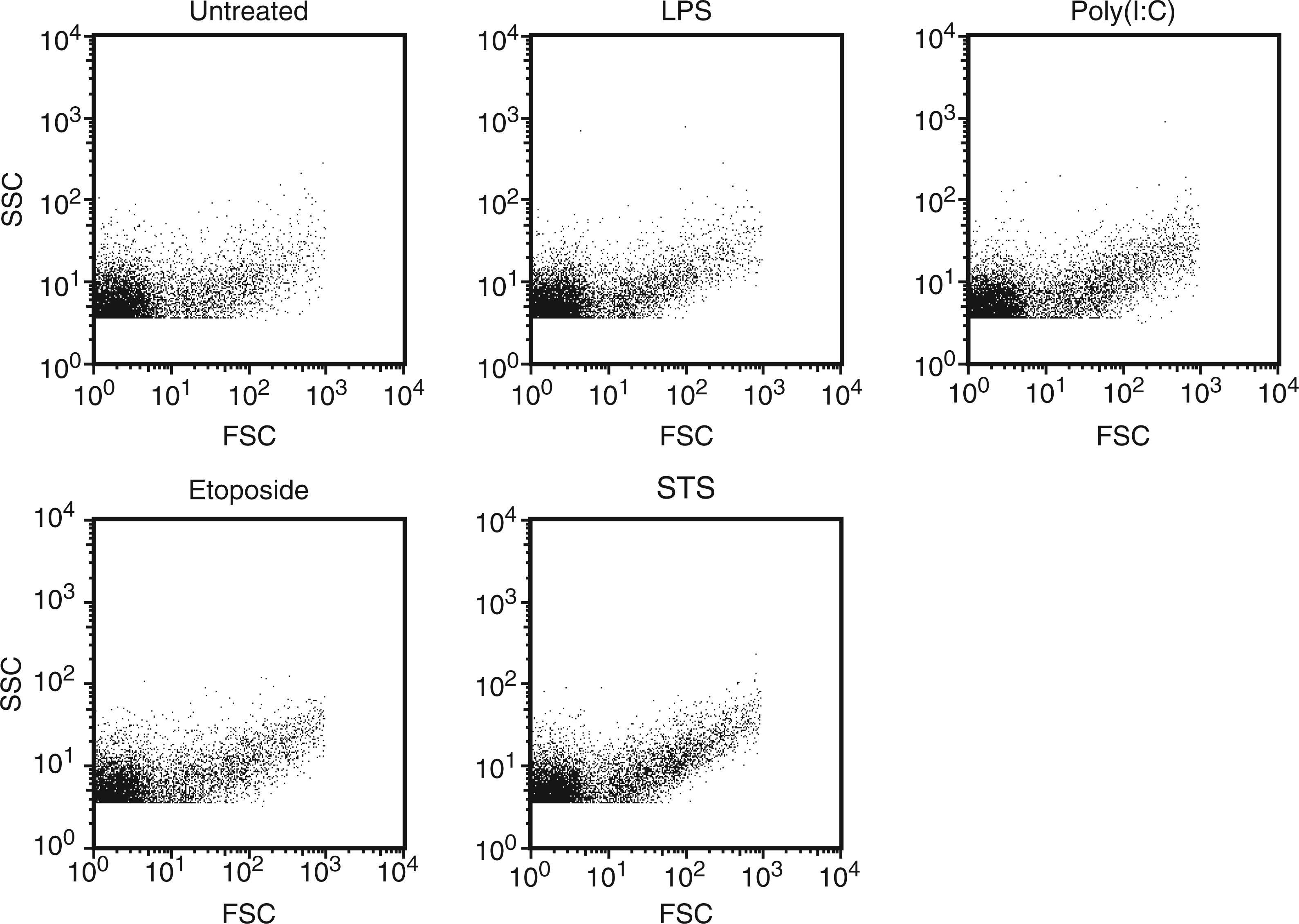
As data shown in Figure 2 indicate, treatment of RAW 264.7 cells to induce activation or apoptosis led to an increase in MP numbers, although the extent varied. Thus, treatment with STS, a protein kinase inhibitor with broad activity, led to much greater MP release than any of the other treatments. In addition, STS caused more extensive release of both DNA and LDH than other treatments, consistent with greater efficacy in inducing apoptosis; these findings do not exclude the possibility that the increase in particles may result from a more direct effect on blebbing or particle release that may not depend on apoptosis.
Analysis of the culture supernatants of RAW 264.7 cells. RAW 264.7 cells were treated for 20–22 h with LPS or poly (I:C) to induce activity with etoposide or STS to induce apoptosis. Cells were then removed by centrifugation. The supernatants were analyzed for numbers of MPs (a), LDH activity (b) and double-stranded DNA concentration (c). Data shown are representative of three separate experiments. Asterisks indicate values significantly different from untreated controls.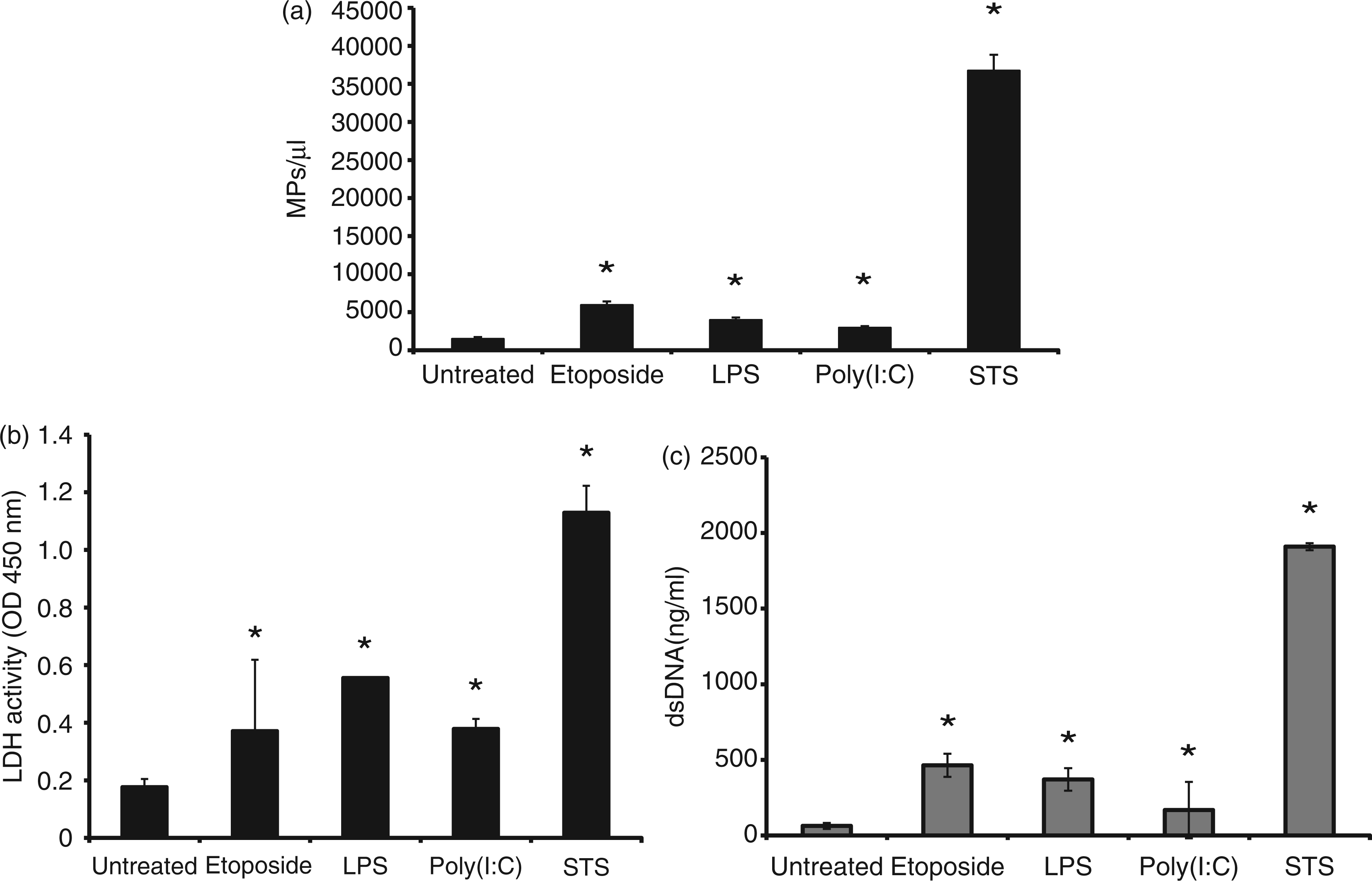
The phenotype of particles from treated and untreated cultures was next analyzed by flow cytometry, assessing annexin V binding as an indicator of the exposure of phosphatidylserine during membrane ‘flipping’, which can be a feature of apoptosis, although it may also occur with certain viable cell populations.21,22 As results presented in Figure 3 indicate, particles treated with etoposide, LPS or poly (I:C) showed similar staining with annexin V as did particles from untreated cultures. The origin of particles in control cultures is difficult to determine, however, as cultures invariably have cells undergoing apoptosis (data not shown). Thus, the control particles may, in fact, represent particles from apoptotic cells.
Annexin V staining of MPs. MPs released by activated, apoptotic and untreated RAW 264.7 cells were stained with FITC-annexin V. The levels of staining were measured by flow cytometry. Data shown are representative of three separate experiments. Black indicates unstained controls. As these data indicate, supernatants from all cultures had particles that bound annexin V, with particles from untreated and LPS-stimulated cells showing a larger percentage of MPs with lower levels of staining.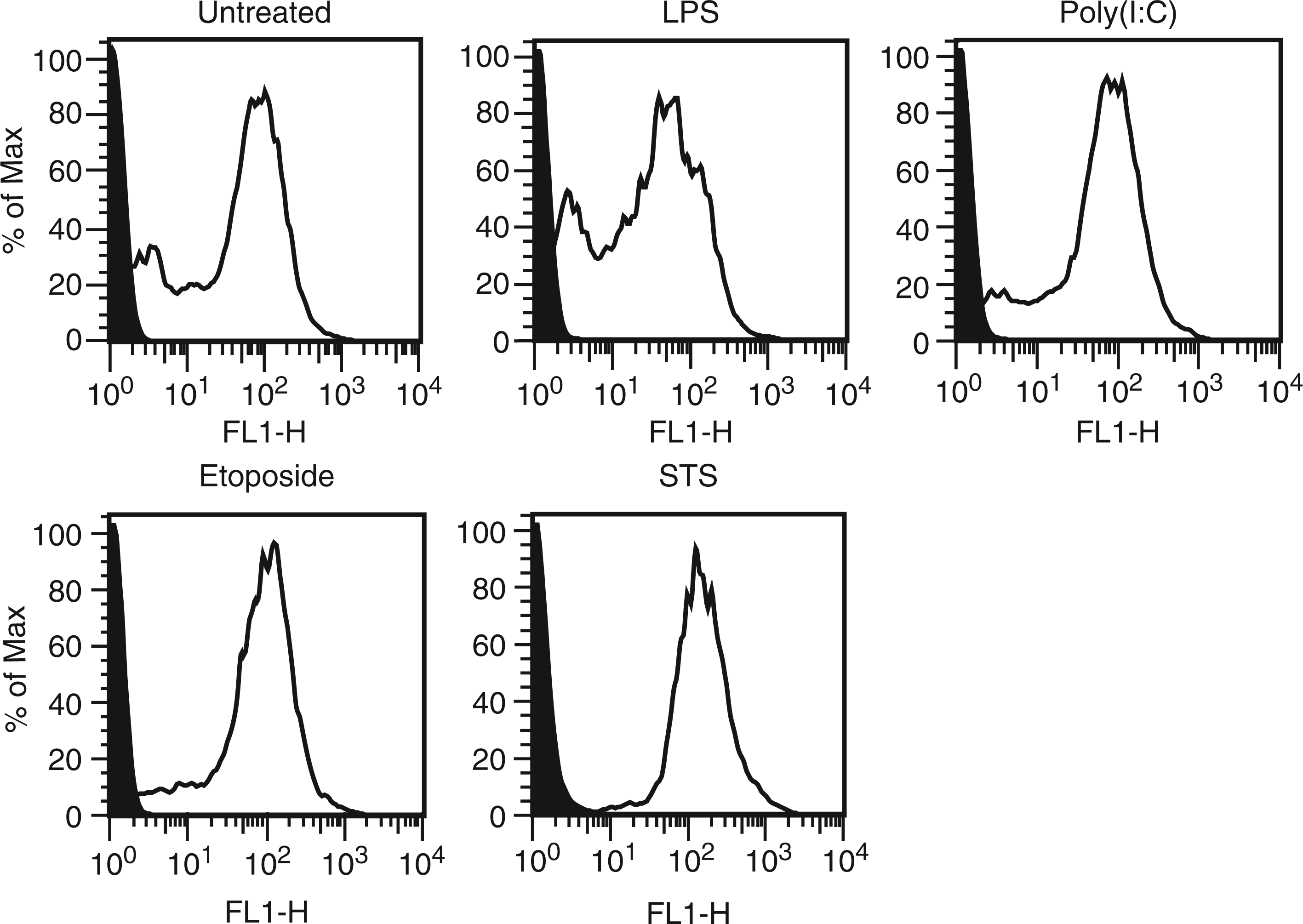
As shown previously in studies of particles from Jurkat T cells and other lymphoid cells undergoing apoptosis, particles can contain both DNA and RNA, as demonstrated by biochemical assays, as well as flow cytometry dyes SYTO 13 and PI, which bind nucleic acids.
19
SYTO 13 can permeate particles irrespective of membrane permeability, while PI enters only those MPs whose membranes are permeable. Figure 4 presents data on the staining of MPs with either SYTO 13 or PI. As these data indicate, MPs stained with SYTO 13 or PI irrespective of source, although there were differences in levels of staining among cell treatments. These differences could relate to MP membrane permeability or the distribution of DNA on the inside or outside of MPs.
Flow cytometry analysis of MPs. MPs released from RAW 264.7 cells treated to undergo activation or apoptosis were stained with either SYTO 13 (a) or PI (b). The stained samples were then analyzed using flow cytometry. Data shown are representative of three separate experiments. MFI: mean fluorescence intensity.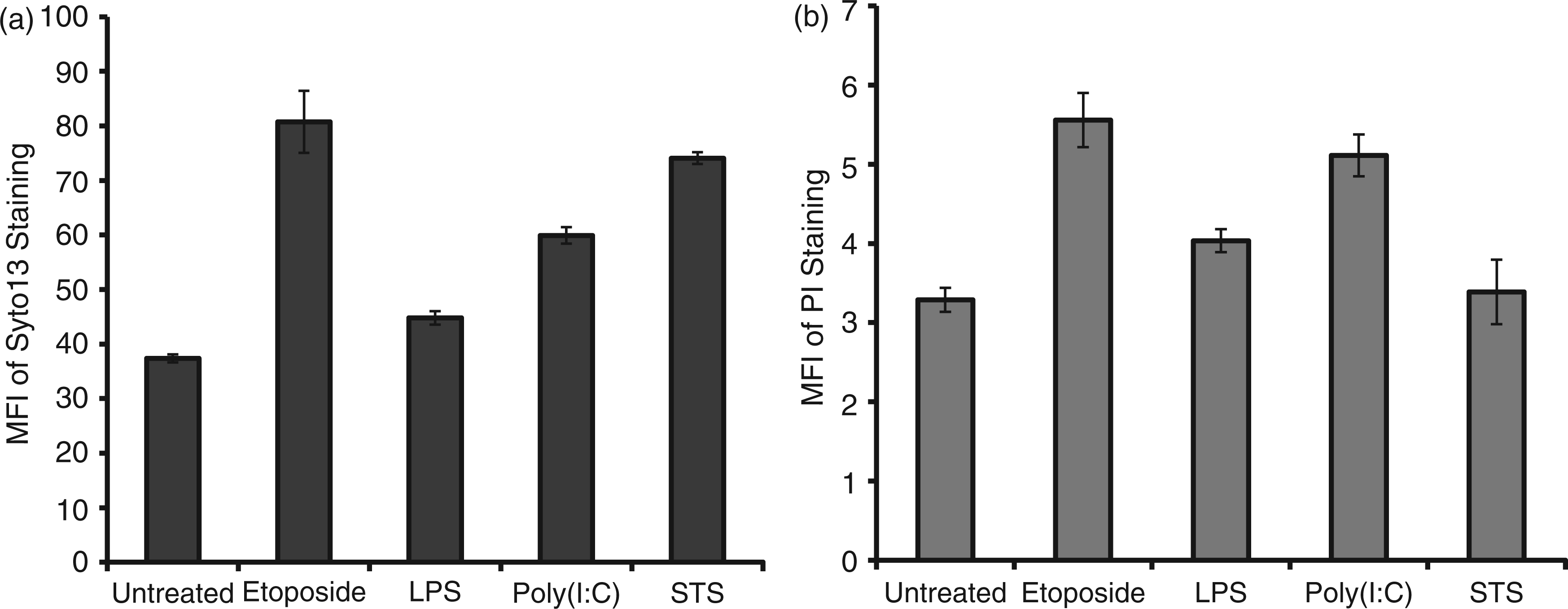
The RNA and content of particles was next analyzed biochemically. Figure 5 presents results of molecular mass determinations of RNA and its integrity as assayed by gel electrophoresis. As results of these studies show, RNA from cells treated with STS differed dramatically from RNA from other particles from either control cultures or cultures treated with LPS, poly (I:C) or etoposide. Thus, the RNA in the particles from STS-treated cultures showed cleavage and degradation, with loss of distinct bands for 18 Sand 28 S rRNA species. In contrast, the RNA from the other cultures showed much less degradation, with distinct bands for 18 S and 28 S rRNA species.
Analysis of MP RNA. RAW 264.7 cells were induced to undergo activation or apoptosis and MPs were prepared. RNA was purified and then examined using an Agilent Bioanalyzer as described in the ‘Materials and methods’ section. The samples shown are RNA from MPs from untreated (B); etoposide (C); LPS (D); STS (E); or poly (I:C) (F) cells. (A) Molecular mass markers. Data shown are representative of three separate experiments.
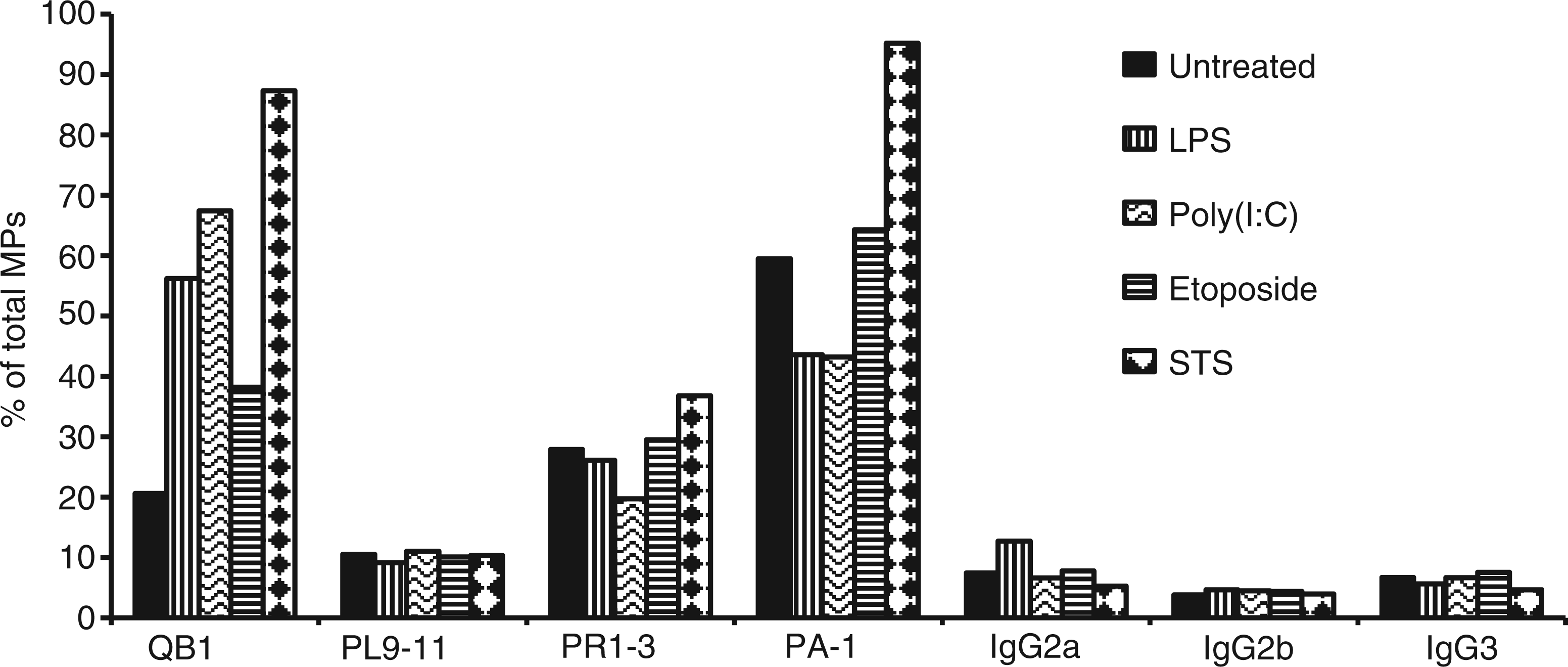
To characterize further the nature of the nucleic acids in particles, we analyzed DNA by gel electrophoresis using preparations isolated using an apoptotic DNA laddering kit. As demonstrated in Figure 6, the preparations from MPs from all conditions were similar, with isolated DNA from all showing evidence of lower molecular mass species and laddering. We next analyzed the DNA content of MPs by flow cytometry using monoclonal Abs that bind to particles from apoptotic cells.19,20 Figure 7 presents these results. As these studies indicate, the monoclonal Abs bound to MPs, independent of source, although particles from STS-treated cultures showed higher levels of staining with Abs QB1 and PAI. These results confirm findings that MPs can display DNA and related nucleosomal molecules in a form accessible to Ab binding.19,20 Taken together with other results, these studies indicate that stimulation of TLRs can elicit particles that are structurally and antigenically similar to those from apoptotic cells.
Analysis of MP DNA by gel electrophoresis. DNA was isolated from purified MPs from RAW 264.7 cells undergoing activation or apoptosis. The samples were analyzed on a 1% agarose gel in TAE buffer at a concentration of 200 ng/lane. The samples were visualized by staining with ethidium bromide. (A) and (G) show molecular mass markers. DNA from MPs from the following cultures were analyzed: (B) untreated; (C) etoposide; (D) LPS; (E) poly (I:C) and (F) STS. Monoclonal Ab binding to MPs. Purified MPs from RAW 264.7 cells undergoing activation or apoptosis were stained with monoclonal Abs recognizing DNA (QB1 and PL9-11) or nucleosomes (PR1-3 and PA-1). The samples were then analyzed by flow cytometry. The data shown are expressed as the percentage of the total population of MPs that bound Ab. The experiment was performed three times. The data shown are representative of the results of all three experiments.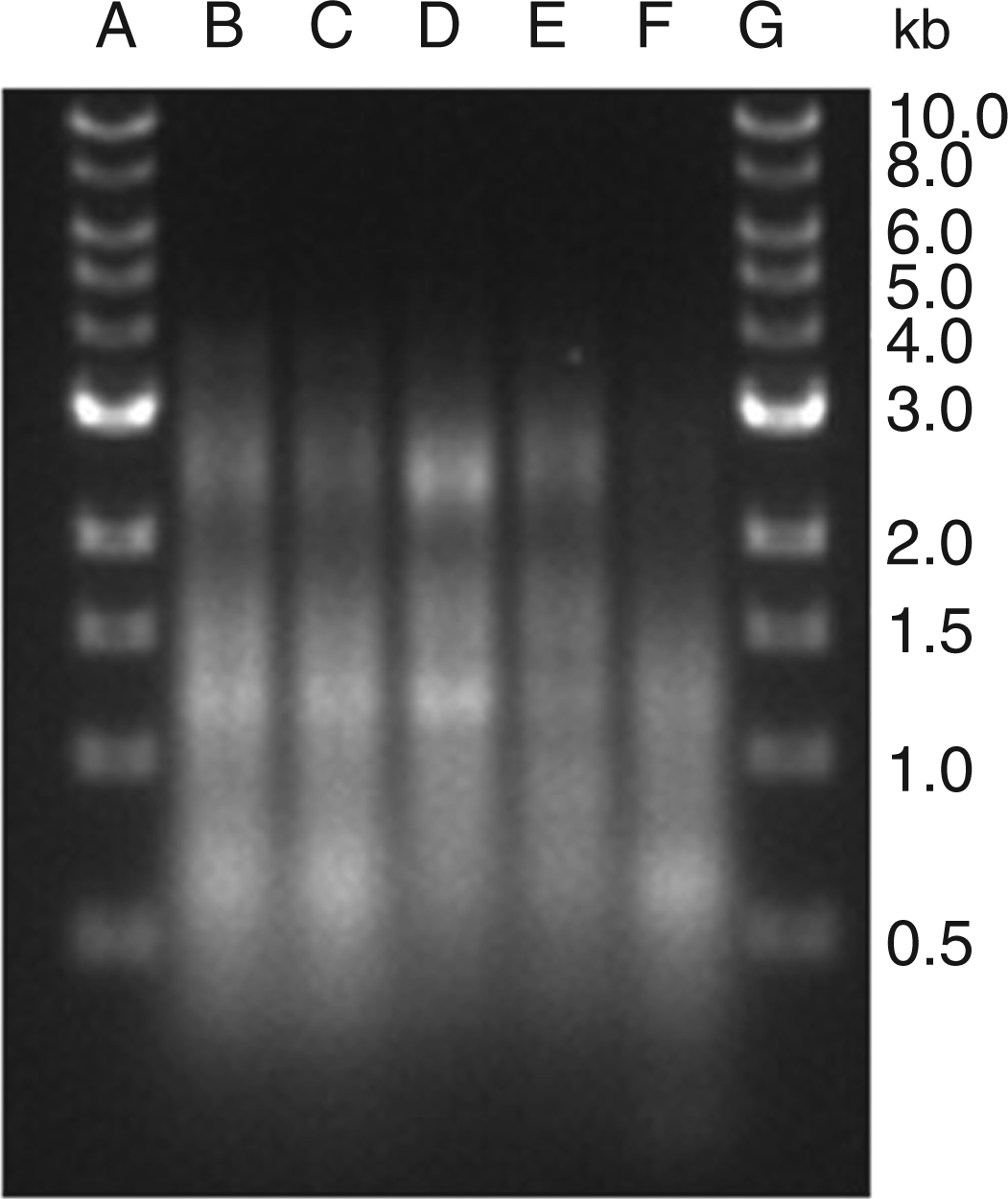
Discussion
Results of these studies provide new insight into the properties of MPs from macrophages. Thus, our studies show that the nucleic acids in MPs from RAW 264.7 cells activated with TLR ligands [LPS or poly (I:C)] resemble those of MPs from apoptotic, as well as untreated, cells. Furthermore, MPs, irrespective of origin, bound monoclonal anti-DNA and related anti-nucleosomal Abs, indicating antigenicity and accessibility of nucleic acids to Abs. As the release of extracellular DNA, in a free or particulate form occurs prominently during cell death,23–25 these findings suggest that MPs released from cells treated with TLR ligands may result from cells undergoing apoptosis during this form of activation. Similarly, MPs from untreated cells may result from apoptosis that occurs during culture of cell lines in vitro.
The translocation of nuclear molecules into blebs is one of the most distinctive features of apoptosis.26–28 While the detailed trafficking of DNA from the nucleus into the cytoplasm has not been defined, blebbing, which leads to MP release, may have an immunological function by compacting DNA into a subcellular structure to promote phagocytosis. This function would be similar to that proposed for apoptotic bodies. Apoptotic bodies are larger than MPs and represent the collapsed remnants of apoptotic cells. 29 The boundary between MPs and apoptotic bodies is not that distinct, however, with size (greater than or less than 1 µm) an important basis of characterization.
The present study extends prior investigations and demonstrates clearly that MPs can contain DNA, as evidenced by biochemical and immunochemical assays, as well as gel electrophoresis. 19 The simplest explanation for this finding relates to the role of LPS and other TLR ligands in inducing cell death. As now recognized, cell death can play an important role in host defense by preventing replication of intracellular organisms and, at the same time, releasing signaling elements (e.g. alarmins and MPs), which can activate other cell types to increase antiviral or antibacterial activity.30–33 This type of death is regulated and programmed, with the immunological outcome determined by downstream pathways induced, including the role of different caspases, as well as extent of cell lysis. Cell death resulting from stimulation of TLR or non-TLR receptors or inflammasome activation may be considered as pyroptosis to signify the subsequent immune stimulation.34,35 This stimulation can result from molecules such as HMGB1 or structures such as MPs.
In in vitro culture systems, it can be difficult to distinguish the relative contributions of cell death and cell activation in the production of MPs in response to TLR stimulation. As we have shown previously, stimulation of particle production by RAW 264.7 cells by LPS is time- and dose-dependent, with the highest number of particles generated at 50 µg/ml—the concentration of LPS used in these experiments. 16 In other studies, we have shown that even lower concentrations of LPS can lead to apoptosis although, under these circumstances, the number of particles is much lower, as is the production of other inflammatory mediators, such as NO. 36 Given the close association of TLR activation and cell death (apoptosis or pyroptosis), determining the origin of a particle from an activated or dying cell in a culture may not be possible.
In view of the potential mechanisms for MP generation, the DNA contained in MPs is most likely nuclear in origin, although mitochondrial DNA may also be present. Mitochondrial DNA is structurally similar to bacterial DNA and, as such, differs from nuclear DNA in its intrinsic immunological activity.37–40 While mitochondria are likely components of MPs, we have not specifically evaluated the content of our particle preparations. In this regard, studies conducted in a variety of species have suggested the existence of another form of DNA called metabolic DNA or the virtosome; this type of DNA may be expendable for the cell and can therefore undergo translocation into the extracellular space without consequences for cell viability. 41 The conditions for the generation and export of metabolic DNA are not well defined, however.
While our studies demonstrate that macrophage MPs contain DNA and related nuclear molecules, the composition of MPs may be heterogeneous and vary depending on their cell of origin or inducing condition. For example, studies have shown that MPs from endothelial cells undergoing stress-induced apoptosis lack DNA and histones, as well as immunostimulatory activity. In contrast, larger structures released from endothelial cells (i.e. apoptotic bodies) contain the nuclear molecules and have pro-inflammatory activity related to expression of IL-1α. 42 These findings suggest that both the translocation of nuclear and the formation of either blebs or apoptotic bodies may reflect cell-specific process that, in turn, affects any subsequent immunes responses.
In addition to their content of DNA, MPs contain RNA of various kinds, including rRNA, mRNA and microRNA (miRNA). The presence of mRNA and miRNA has suggested a role of MPs in intracellular communication by transfer of informational RNA. Our previous studies showed that rRNA from particles obtained from Jurkat and HL-60 cells undergoing apoptosis induced by STS, UV treatment or camptothecin showed significant degradation. 19 In addition, we showed that MP rRNA differed from that in the apoptotic cells in the extent of the cleavage. These findings suggested either selective incorporation of degraded RNA into particles or distinct patterns of RNA degradation in the particles compared to cells. As in studies by others, cleavage of rRNA can be a feature of apoptotic death.43–46
The results of the current study indicate differences in the pattern of RNA degradation of the particles from RAW 264.7 cells depending on the inducing stimulus. Whereas particles from cells treated with STS showed extensive RNA degradation, RNA from particles from cells treated with either LPS, poly (I:C) or etoposide showed much more intact RNA, with clear bands expected for 28 S and 18 S species visible. The differences in the patterns of rRNA integrity could result from the kinetics and extent of cell death depending on the inducing agent; in our experiments, STS led to the highest level of cell death, as shown by the extent of LDH production and can induce cell death rapidly (preliminary observations). The pattern of RNA degradation may relate to another action of STS in RAW 264.7 cells that is not directly related to apoptosis. Staurosporine is a broad spectrum kinase inhibitor and can also affect blebbing among many other processes. 47
Among their other properties, MPs display nuclear antigens that can serve as a target of auto-Abs in the setting of systemic lupus erythematosus, a prototypic autoimmune disease characterized by immune complex formation.20,48 In our previous studies, we showed that monoclonal Abs to DNA and other nucleosomal antigens can bind to particles from apoptotic cells and that particles from patients with lupus can have bound IgG, indicative of immune complex formation. 20 Our current studies indicate that particles from activated cells are also auto-antigenic. Thus, in the setting of autoimmune disease, the presence of particles with bound IgG is consistent with either an event (e.g. ischemia, UV damage) that leads primarily to apoptosis or an event resulting from intense immune activation or sterile inflammation. As the particles resulting from these processes can have similar properties, it may be difficult to distinguish proximal events leading to their production. Future studies will determine whether the functional properties of the particles vary depending on mode of induction and content of other bioactive molecules.
Footnotes
Funding
This work was supported by a VA Merit Review Grant, a grant from Alliance for Lupus Research (ALR) and NIH 5U19-AI056363.