
Abstract
The interaction between surfactant protein-A (SP-A) and TLR4 is important for host defense. We have recently identified an SPA4 peptide region from the interface of SP-A–TLR4 complex. Here, we studied the involvement of the SPA4 peptide region in SP-A–TLR4 interaction using a two-hybrid system, and biological effects of SPA4 peptide in cell systems and a mouse model. HEK293 cells were transfected with plasmid DNAs encoding SP-A or a SP-A-mutant lacking SPA4 peptide region and TLR4. Luciferase activity was measured as the end-point of SP-A–TLR4 interaction. NF-κB activity was also assessed simultaneously. Next, the dendritic cells or mice were challenged with Escherichia coli-derived LPS and treated with SPA4 peptide. Endotoxic shock-like symptoms and inflammatory parameters (TNF-α, NF-κB, leukocyte influx) were assessed. Our results reveal that the SPA4 peptide region contributes to the SP-A–TLR4 interaction and inhibits the LPS-induced NF-κB activity and TNF-α. We also observed that the SPA4 peptide inhibits LPS-induced expression of TNF-α, nuclear localization of NF-κB-p65 and cell influx, and alleviates the endotoxic shock-like symptoms in a mouse model. Our results suggest that the anti-inflammatory activity of the SPA4 peptide through its binding to TLR4 can be of therapeutic benefit.
Introduction
An uncontrolled inflammatory response against infectious stimuli can lead to severe damage or failure of important organs. Endotoxic shock-induced acute respiratory distress syndrome (ARDS) and multiple organ failure represent this condition.1,2 Conventional corticosteroids and selective blockade of isolated aspects of inflammatory state are practised; none of them have been proven completely beneficial for the treatment of ARDS. 3 Corticosteroids are known to affect functions of many cells and systems within the body, including the suppression of the immune system. Others target the preformed, already-secreted cytokines and chemical mediators, for example anti-TNF-α, IL-1β and IL-6-Abs, and receptor antagonists. These agents provide only a transient relief and have significant side effects, including a serious risk of secondary infections.4,5 Therapeutic inhibition of inflammatory signaling may provide better results and help alleviate the clinical symptoms.
Toll-like receptor-4 (TLR4) recognizes pathogen-associated molecular patterns derived from pathogens, including the LPS of Gram-negative bacteria. It also recognizes endogenous damage-associated molecular patterns, for example fibronectin and heat-shock proteins, released as a consequence of an inflammatory response.
6
Upon binding to the ligand, TLR4 induces a complex intracellular signaling through its Toll/interleukin-1 receptor domain and transcription factors, including NF-κB, which leads to synthesis of cytokines/chemokines and other inflammatory mediators. Surfactant protein-A (SP-A) expressed by epithelial cells at various mucosal surfaces in our body is recognized as the secretory pathogen recognition receptor.
7
We, and others, have shown that SP-A interacts with multiple receptors, including TLR4.8–12 Through interaction with TLR4, the SP-A modulates host defense functions of immune cells against infectious stimuli: SP-A reduces the release of TLR4-stimulated pro-inflammatory TNF-α cytokine, but preserves TLR4-induced phagocytosis.12,13 Thus, a TLR4-interacting region of SP-A may mimic host defense functions of SP-A. We have recently identified a 20-mer SPA4 peptide with amino acid sequences, G
Our results demonstrate that the SPA4 peptide region contributes to SP-A–TLR4 interaction and inhibits LPS-induced inflammatory parameters—TNF-α secretion, NF-κB activity, influx of cells—and alleviates the endotoxic shock-like symptoms in a mouse model of LPS-induced inflammation.
Materials and methods
Animals
The animal studies were approved by the Institutional Animal Care and Use and Institutional Biosafety Committees at the University of Oklahoma Health Sciences Center (OUHSC), Oklahoma City, OK, USA. BALB/c mice (female, 5–6 wk old) were included and housed for 1 wk for acclimatization at the College of Pharmacy Animal Facility, OUHSC prior to conducting any experiment.
Protein–protein interaction by mammalian two-hybrid assay
Interaction between (i) SP-A and TLR4, and (ii) an SP-A-mutant lacking SPA4 peptide region and TLR4, was assessed in HEK293 cells by a mammalian two-hybrid assay (Promega, Madison, WI, USA). The functional relevance of the SP-A–TLR4 interaction was studied simultaneously by measuring NF-κB activity in this system.
HEK293 cell culture
We included human embryonic kidney (HEK) epithelial cells (HEK293, obtained from Dr Kelly Standifer, Department of Pharmaceutical Sciences, OUHSC) to study the protein–protein interaction because HEK293 cells do not express endogenous SP-A or TLR4 (Figure 1). The HEK293 cells were maintained in DMEM containing 0.37% sodium bicarbonate, 5% heat-inactivated FBS, 100 U/ml penicillin and 100 µg/ml streptomycin antibiotics (Invitrogen, Grand Island, NY, USA).
(A) (i) Expression of SP-A and SP-A-mutant proteins as fusion proteins with VP16, and TLR4-GAL4 fusion protein by transiently-transfected HEK293 cells. Ten micrograms of total cell lysate proteins were separated on 4–20% Tris-glycine SDS-PAGE gradient gel under complete reducing (heating for 5 min + 11 mM β-mercaptoethanol) or partially-reducing (heating for 5 min) conditions and probed with SP-A- or TLR4-specific antibody (Ab), respectively. Results in the left panel indicate SP-A–Ab reactive bands. Lane 1: purified lung SP-A (10 ng); lane 2: lysate protein from cells transfected with pSPA-mutant and pTLR4; lane 3: lysate protein from cells transfected with pSPA and pTLR4 plasmid DNA constructs; and lane 4: lysate protein from non-transfected cells. Results in the right panel demonstrate the TLR4-Ab reactive bands. Lane 1: lysate protein from cells transfected with pSPA-mutant and pTLR4; lane 2: lysate protein from cells transfected with pSPA and pTLR4 plasmid DNA constructs; and lane 3: lysate protein from non-transfected cells. (ii) Secreted amounts of SP-A in cell-free supernatants of non-transfected, pSPA + pTLR4 and pSPA-mutant + pTLR4 co-transfected HEK293 cells. SP-A levels (in ng) normalized with total µg cellular proteins are listed. (iii) Cell-surface expression of TLR4 in non-transfected HEK293 cells, and in pSPA + pTLR4 and pSPA-mutant + pTLR4 co-transfected HEK293 cells. Percent number of TLR4-positive cells (T) in region R is shown within the histogram plots. Isotype control Ab-stained cells were included as control (IC). (iv) Confocal microscopy of the SP-A- and TLR4-Ab stained non-transfected HEK293 cells (negative control). Non-transfected HEK293 cells were permeabilized, immunostained with Abs against SP-A (green) and TLR4 (red), and counterstained with nuclear dye (blue). Scale bar is shown within the image. (v) Expression of SP-A (in green) and TLR4 proteins (in red), and their co-localization (in yellow) in HEK293 cells transfected with pSPA and pTLR4 by confocal microscopy. Cells were permeabilized and immunostained with Abs against SP-A and TLR4 proteins, and counterstained with nuclear dye (blue). The confocal images in the bottom panel are from a single HEK293 cell transfected with pSPA and pTLR4 plasmid DNA constructs. (B) Loss of SPA4 peptide region in SP-A-mutant protein results in decreased Firefly luciferase activity in cells transfected with pSPA-mutant and pTLR4. RLU depicting interaction between SP-A and TLR4 (100) and SP-A-mutant protein and TLR4 by two-hybrid assay. Negative controls included HEK293 cells transfected with pACT and pBIND (vector backbones). Additional controls included were cells transfected with pSPA, pBIND and pcDNA3.0 and non-transfected cells (not shown). Positive control included cells transfected with pACT-MyoD and pBIND-ID plasmid DNA constructs (not shown). The error bars represent SEM. Results are from four experiments performed separately at different times. Statistical significance (P-value < 0.007) is shown as compared to SP-A–TLR4 interaction (t-test). (C) The NF-κB luciferase reporter activity was measured in HEK293 cells transfected with pSPA or pSPA-mutant and pTLR4 plasmid DNA constructs, and challenged with LPS (100 ng/ml) for 5 h. The Renilla luciferase activity was measured to assess the transfection efficiency. The luminescence values for NF-κB-associated Firefly luciferase activity normalized with those for Renilla luciferase activity are shown as bar chart. The bars show mean + SEM of results from three separate experiments performed at different times. P < 0.001 was noted as compared to the values in cells transfected with pTLR4 only (ANOVA).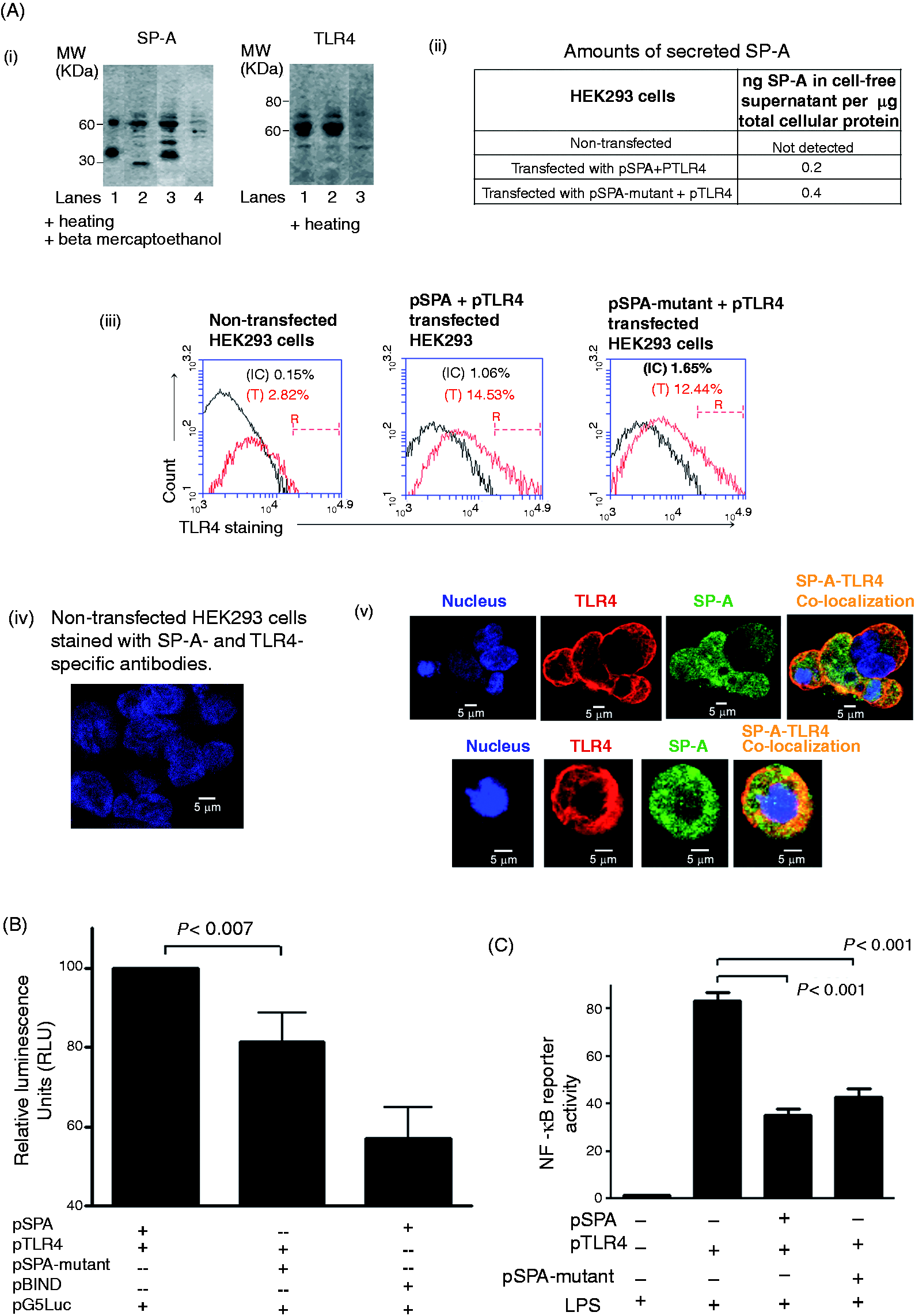
Plasmid DNA constructs
Plasmid DNA constructs encoding full-length human SP-A (pSPA), SP-A-mutant lacking SPA4 peptide region (pSPA-mutant) and full-length human TLR4 (pTLR4) were prepared using recombinant DNA methods (Mutagenex, Piscataway, NJ, USA). The SP-A– and TLR4–cDNA inserts were obtained from pCR-BluntII-TOPO (Open Biosystems, Lafayette, CO, USA) and pUNO1-hTLR04a (Invivogen, San Diego, CA, USA) plasmid DNAs, respectively. SalI and MluI restriction sites were added to the SP-A–cDNA fragment and cloned into the pACT plasmid DNA to obtain pSPA (Promega). Subsequently, the pSPA-mutant was prepared by site-directed mutagenesis. The pTLR4 construct was prepared by PCR subcloning of the TLR4-cDNA insert into the pBIND plasmid DNA backbone at the BamHI-MluI sites (Promega). The pSPA and pSPA-mutant plasmid DNA constructs were designed to encode SP-A and SP-A-mutant proteins as fusion proteins with VP16, and pTLR4 plasmid construct to encode TLR4 as fusion protein with GAL4, respectively. The cloning direction, reading frame and insert size within the plasmid constructs were confirmed by DNA sequencing and restriction digestion analysis. The plasmid DNAs were prepared using endotoxin-free plasmid DNA extraction kit (Qiagen, CA), and endotoxin content was checked by Limulus amebocyte lysate (LAL) assay kit (Charlesriver Lab, MA).
Mammalian two-hybrid assay
The cells were transfected with pSPA or pSPA-mutant, pTLR4 and pG5Luc (encoding Firefly luciferase) plasmid DNAs. Transfection conditions were optimized using different cell numbers and plasmid DNAs to Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) ratios. In the comprehensive experiments, HEK293 cells were seeded at a density of 2.5 × 105 cells per ml and transfected using 0.2 µg plasmid DNA each per 1 µl Lipofectamine 2000 transfection reagent. After 24 h of transfection, the cells were washed and scraped in 20 µl lysis buffer provided with the Dual Luciferase Reporter Assay Kit (Promega). Firefly and Renilla luciferase-associated luminescence were read using the Synergy HT multi-mode microplate reader (Biotek, Winooski, VT, USA). The lysates of (i) non-transfected cells and cells transfected with (ii) pACT and pBIND vector plasmid DNAs, and (iii) pSPA and pBIND (vector backbone for pTLR4), served as negative controls. Plasmid DNAs (pACT-MyoD and pBIND-Id) provided with the kit, served as assay controls.
The following calculations were performed after obtaining the raw data for Firefly and Renilla luciferase. 14 The Firefly luciferase activity-associated luminescence value for each cell lysate was divided by its Renilla luciferase activity-associated luminescence value and multiplied by 1000. The luciferase activity was then expressed in relative luminescence units (RLU). In all the experiments, the RLU for cells transfected with pSPA and pTLR4 was set at 100.
Immunoblotting for SP-A and TLR4
The cells were homogenized in a homogenization buffer containing a cocktail of protease inhibitors (1 mM EDTA, 1.1 µM leupeptin, 1 µM pepstatin, 0.2 mM phenylmethyl sulphonyl fluoride) and detergents (0.1% SDS, 1% Igepal CA630). 15 Total protein concentration was measured in lung tissue homogenates by bicinchoninic acid (BCA) protein assay kit. The total cell lysate proteins were separated on Novex 4–20% tris-glycine SDS-PAGE gradient gel (Invitrogen, Carlsbad, CA, USA) and transferred onto nitrocellulose membrane. The non-specific sites were blocked using 7% non-fat milk solution. The membrane was then incubated with 1:1000 diluted anti-human SP-A or 1:500 diluted anti-human TLR4 (Abcam, Cambridge, MA, USA) Abs. The membrane was washed and incubated further with 1:1000 diluted anti-rabbit HRP-conjugated Ab for 45 min. The immune complexes were visualized using the chemiluminescent substrate reagent (Pierce, Rockford, IL, USA). Immunoblots were imaged using the Ultraquant Acquisition program (Ultralum, Claremont, CA, USA). Purified human lung SP-A (provided by Dr Jo Rae Wright, Department of Cell Biology, Duke University Medical Center, Durham, NC, USA) was included as positive control for SP-A.
ELISA for SP-A
The secreted levels of SP-A were measured in freeze-concentrated cell-free supernatants by ELISA as per the method described earlier. 16 Briefly, the cell-free supernatants diluted in 0.1 M NaHCO3 buffer at pH 9.6 were incubated overnight in multi-well Immulon strips (Thermo Electron Corp, Milford, MA, USA). The non-specific sites were blocked, and 1:1000 diluted rabbit anti-human SP-A specific Ab was added to the wells. After washing the wells, the immune complexes were incubated with 1:1000 diluted secondary anti-rabbit HRP-conjugated Ab following the incubation with 75 µl of 3,3’,5,5’-tetramethylbenzidine substrate solution. The reaction was stopped using 0.2 N H2SO4. The optical density was read spectrophotometrically. Diluted amounts of purified human lung SP-A were used to prepare the standard curve. Measured amounts of SP-A were normalized with total cellular protein.
Immunocytochemistry
To confirm the expression and co-localization of SP-A and TLR4 proteins in transfected cells, the immunocytochemistry was performed in 8 well chamber slides (Nunc, Rochester, NY, USA). Non-specific sites were blocked with 1% BSA and cells were incubated with 1:500 diluted anti-human SP-A (Chemicon, Billerica, MA, USA) and 200 µg/ml anti-human TLR4 (Imgenex, San Diego, CA, USA) Abs for 17 h (overnight) at 4°C. Subsequently, cells were washed and incubated with Alexa fluor 488-conjugated anti-rabbit Ab for SP-A and Alexa fluor 568-conjugated anti-mouse Ab for TLR4 (10 µg/ml; both Abs were from Invitrogen, Carlsbad, CA, USA). Finally, Hoechst dye (1 µg/ml) was added for nuclear staining and the slides were mounted with Vectashield (Vector Laboratories, Burlingame, CA, USA). All the images were acquired at 63 × oil immersion objective under Zeiss confocal microscope and processed using Zeiss LSM Image Examiner or ZEN 2011 programs.
Flow cytometry
The cell surface expression of TLR4 was also investigated by flow cytometric analysis of cells transfected with pSPA or pSPA-mutant, pTLR4 and pG5Luc plasmid DNAs. After 24 h of transfection, cells were washed twice with ice-cold Dulbecco’s PBS (DPBS), stained with phycoerythrinin-conjugated anti-TLR4 Ab (clone HTA125; eBioscience, San Diego, CA, USA) for 45 min and fixed in 0.5% paraformaldehyde solution. Finally, the fixed cells were run on Accuri flow cytometer (BD Biosciences, San Jose, CA, USA), and TLR4-staining was analyzed using C6 software.
NF-κB reporter activity assay
The functional relevance of interaction of SP-A or SP-A-mutant with TLR4 was assessed in the two-hybrid HEK293 cell system by NF-κB reporter activity assay. Briefly, HEK293 cells (50,000 per well) were seeded in a 96-well tissue-culture plate (BD Falcon, Franklin Lakes, NJ, USA). The cells were transfected with 0.2 µg plasmid DNA each of pSPA or pSPA-mutant, pTLR4 and pGL4.32 NF-κB reporter plasmid DNA (luc2P/NF-κB-RE/Hygro; Promega) using 1 µl Lipofectamine 2000 reagent per well. After 24 h of transfection, cells were washed once with plain DMEM and treated with highly purified, low lipoprotein Escherichia coli O111:B4 LPS (100 ng/ml; Calbiochem, La Jolla, CA, USA) for 5 h. After completion of incubation, cells were washed once with ice-cold DPBS and scraped in 20 µl of cell lysis buffer provided with the Dual Luciferase Reporter Assay Kit (Promega). Firefly and Renilla luciferase readings were recorded using the Synergy HT multi-mode microplate reader (Biotek). Renilla luciferase activity provided a measurement of transfection efficiency. The luminescence units obtained for NF-κB-associated Firefly luciferase activity were normalized with those readings for Renilla luciferase. The pTLR4-transfected cells challenged with LPS alone served as control.
Synthetic SPA4 peptide
After assessing the role of SPA4 peptide region in SP-A-TLR4-interaction and function in HEK293 two-hybrid assay system, the 20-mer SPA4 peptide (amino acid sequence: GDFRYSDGTPVNYTNWYRGE) was included to investigate its direct anti-inflammatory effects in a dendritic cell line and in a mouse model. The SPA4 peptide was synthesized by Genscript (Piscataway, NJ, USA) and purity was confirmed by mass spectroscopy and HPLC. Endotoxin content in SPA4 peptide suspensions was measured by Limulus amebocyte lysate (LAL) assay. Predictions about primary structure, physico-chemical features and three-dimensional (3D) confirmation were obtained using PepDraw (Tulane University, New Orleans, LA, USA; 17 www.tulane.edu/∼biochem/WW/PepDraw/index.html) and PEP-FOLD programs, respectively. 18 A Kyte and Doolittle hydropathy plot was drawn to determine the hydrophobicity/hydrophilicity of the SPA4 peptide. 19
SPA4 peptide activity at cellular level
As HEK293 cells do not represent the immune antigen-presenting cells, the biological effects of synthetic SPA4 peptide were studied in a dendritic cell system.
JAWS II dendritic cell culture
The JAWS II dendritic cells (ATCC, Manassas, VA, USA) derived from the bone marrow of C57BL/6 mice were maintained in alpha-modified minimum essential medium (α-MEM; Sigma, St. Louis, MO, USA) supplemented with 20% FBS, 4 mM
Expression of phospho-NF-κB-p65
The JAWS II dendritic cells (1 × 106 cells) were challenged with highly-purified, low lipoprotein E. coli O111:B4 LPS (100 ng/ml; Calbiochem) for 4 h and subsequently treated with SPA4 peptide (1 and 10 µM) for a period of 1 h. The cell lysates were prepared in 200 µl homogenization buffer containing protease inhibitors, detergents (as described above) and phosphatase inhibitors (0.25 mM sodium orthovandate and 500 mM sodium fluoride). The cell lysate proteins were separated on 4–20% Tris-glycine SDS-PAGE gel under complete reducing conditions. The expression of phosphoryated-NF-κB-p65 (Ser 276) was investigated by immunoblotting with 1:500 diluted anti-phospho-NF-κB-p65 Ab (Santacruz Biotechnology, Santa Cruz, CA, USA) and 1:2000 diluted anti-rabbit HRP-conjugated secondary Ab, as per the method described above. The membrane was stripped off the probing Abs at 60°C for 45 min using a stripping solution containing 10% SDS, 0.5 M Tris and β-mercaptoethanol (35 µl per ml) and re-probed with 1:1000 diluted anti-β-actin Ab (Biolegend, San Diego, CA, USA) to confirm equal loading. Immunoblots were imaged using the Ultraquant Acquisition program (Ultralum), and densitometric analysis of immunoreactive bands was performed with the Image J 1.42q program. Finally, the arbitrary densitometric units for the phospho-NF-κB-p65 were normalized with those for β-actin.
NF-κB activity assay
The JAWS II cells (1 × 106 cells) were co-transfected with pcDNA3.0 vector (obtained from Dr Brian Ceresa, Department of Cell Biology, OUHSC) or myeloid differentiation primary response gene (MYD88)-dominant negative (MYD88DN) lacking the death and intermediate domains (obtained from Dr. Ruslan Medzhitov, Yale University, CT) and pGL4.32 NF-κB reporter plasmid DNA (luc2P/NF-κB-RE/Hygro; Promega) plasmid DNA constructs (1 µg DNA each) using TransIT-TKO transfection reagent (Mirus, Madison, WI, USA).21,22 The ratio of transfection reagent to DNA (2 µl per 1 µg of DNA) was kept constant. After 4 h of incubation, cells were supplemented with an additional 250 μl of α-MEM medium containing 20% plain FBS and incubated for an additional 14–16 h. Cells were then washed and treated with highly purified, low lipoprotein E. coli O111:B4 LPS (100 ng/ml; Calbiochem) for 4 h and subsequently treated with SPA4 peptide (1 and 10 µM) for a period of 1 h. Luminescence of the NF-κB-associated Firefly luciferase activity was read using the Synergy HT multi-mode microplate reader, as described earlier. Total cellular protein content was estimated using a BCA protein assay kit (Pierce) and was used to normalize the NF-κB-associated luciferase activity.
Cytokine (TNF-α) measurement
The TNF-α levels were measured in cell-free supernatants of JAWS II cells treated with LPS ± SPA4 peptide by ELISA. 23 The secreted levels of TNF-α were normalized with total cellular protein.
Mouse model of LPS-induced lung inflammation
A mouse model of LPS-induced lung inflammation was included to assess the in vivo efficacy of SPA4 peptide. In the pilot experiments, mice were injected with 1.0, 10, 15 and 20 µg of LPS per g body mass intraperitoneally (IP), and observed for endotoxic shock-like symptoms and histological evidence of an influx of leukocytes. The dose of 15 µg LPS/g body mass was found to be optimum to induce endotoxic shock-like symptoms and severe inflammation in lung without causing any mortality within the study period. Thus, for the comprehensive experiments, we challenged the mice with 15 µg LPS/g body mass. Control mice received an equal volume of endotoxin-free saline. After 1 h of LPS challenge, mice were injected with SPA4 peptide: 2.5 µg/g body mass or purified lung SP-A: 0.5 µg/g body mass (provided by Dr Jo Rae Wright, Department of Cell Biology, Duke University Medical Center, Durham, NC, USA) IP. The treatment dose of SPA4 peptide was kept five times higher than that of purified lung SP-A because of the difference in their binding affinity to TLR4. 12
Mice were monitored for signs of endotoxic shock-like symptoms. After 6 h of LPS-challenge, the endotoxic shock-like symptoms (ruffled fur, eye exudates, prostration, signs of diarrhea and lack of reactivity) were noted for each mouse on a scale of 0–3. An average symptom index score was obtained for each mouse in a group. 24
Subsequently, mice were anesthetized and euthanized. A sample of blood was collected via cardiac puncture, centrifuged to obtain serum, aliquoted and stored at −80°C. Major organs were harvested under aseptic conditions. Lung tissues were either snap-frozen in liquid nitrogen or fixed in 10% buffered formalin. At the time of analyzing inflammatory parameters, frozen lung tissues were thawed and homogenized in a homogenization buffer containing a cocktail of protease inhibitors and detergents, as described earlier. 15 Total protein concentration was measured in lung tissue homogenates by a BCA protein assay kit.
Tissue histopathology
After fixing overnight in 10% buffered formalin, the lung tissue specimens were transferred to 75% ethanol. Tissues were processed to further dehydrate, clear and infiltrate into 70–100% alcohol and xylene, and embedded into paraffin. Lung sections (5 µm in thickness) were obtained and stained with hematoxylin and eosin (H&E).
Measurement of cytokine (TNF-α)
The TNF-α levels were measured in diluted serum samples and lung tissue homogenates by ELISA. 21 The amounts of TNF-α measured in lung homogenates were normalized with total protein.
Levels of myeloperoxidase in lung tissue homogenates
Myeloperoxidase (MPO; EC 1.11.1.7) is a lysosomal hemeprotein located in the azurophilic granules of neutrophils and monocytes. Increased MPO expression is associated with inflammation. 25 Thus, we measured the MPO levels in lung homogenates using a commercially-available ELISA kit (Invitrogen–Molecular Probes, Carlsbad, CA, USA). Briefly, plate wells were coated with 500 ng/ml mouse anti-MPO Ab for 1 h at 22–24°C (room temperature). After washing the wells, MPO standard solutions (0.75–100 ng/ml) and diluted lung homogenates were added and incubated for 1 h at room temperature. The antigen (Ag)–Ab immune complexes were then incubated with 1 µg/ml rabbit anti-MPO secondary capture Ab and 100 ng/ml goat anti-rabbit HRP-labeled IgG. Finally, the Amplex UltraRed reagent, a fluorogenic substrate for HRP, was added and fluorescence was read at the setting of 530 nm (excitation) and 590 nm (emission) wavelengths on the Synergy HT multi-mode microplate reader.
Lung immunohistochemistry for NF-κB-p65
Five-micrometer sections of lung were de-paraffinized in xylene and rehydrated through a graded ethanol series. Ag retrieval was performed in pH 6.0 citrate buffer prior to staining. Non-specific binding sites were blocked with normal mouse serum. The tissue sections were then incubated overnight with NF-κB-p65 Ab (1:5000 dilution, Santa Cruz Biotechnology). Finally, NF-κB-p65 localization was detected using Vectastain ABC anti-rabbit IgG kit and Vector Blue alkaline phosphatase substrate system (Vector Laboratories). The lung sections were counterstained with nuclear fast red, cleared with a xylene substitute, and coverslips were mounted permanently using non-xylene based mounting medium. The slides were examined for NF-κB-p65 expression and nuclear localization under light microscope.
Statistical analysis
The results were analyzed for statistical significance by Student’s t-test or ANOVA using Prism software (Graphpad, La Jolla, CA, USA). The P-values at < 0.05 were considered significant or otherwise noted.
Results
In our earlier study, we identified the SPA4 peptide region from the TLR4-interacting site in an in silico model of SP-A–TLR4–MD2 complex. Direct binding of the synthetic SPA4 peptide to TLR4 was studied using an in vitro microwell-binding assay. 12 Relevance of the SPA4 peptide region in SP-A-TLR4-interaction remained unknown in cellular system. Here, we utilized a mammalian two-hybrid assay to assess the contribution of SPA4 peptide region in the SP-A–TLR4 interaction in HEK293 cells, which do not express endogenous SP-A or TLR4, and provide a cleaner system for analysis of SP-A–TLR4 interaction. The two-hybrid system is based on the modular domains found in some transcription factors: a DNA-binding (DB)-domain, which binds to a specific DNA sequence, and a transcriptional activation (TA)-domain, which interacts with the basal transcriptional machinery. A TA-domain in association with a DB-domain promotes the assembly of RNA polymerase II complexes at the TATA box and increases transcription. The DB-(GAL4) and TA-(VP16) domains are produced by separate plasmids. 26 The two-hybrid assay has been utilized for studying interaction between various protein partners.27,28 Similarly, the transcription machinery was expected to be activated by interaction between TLR4 fused to a GAL-4 DB-domain and SP-A protein fused to a VP16 TA-domain. The SP-A-TLR4 interaction would then result in the transcription of Firefly luciferase reporter gene.
We utilized HEK293 cells because the HEK293 cells did not express endogenous SP-A or TLR4 proteins as confirmed by real-time PCR (results not shown), Western blotting, flow cytometry and immunocytochemistry (Figure 1A). All the experiments were performed in endotoxin-free conditions; the plasmid DNA suspensions had < 0.000285 ng endotoxin per µg DNA, as detected by LAL assay kit. Under optimized experimental conditions, we obtained about 70% transfection efficiency, as assessed by visualizing the green fluorescence in cells transfected with pHYG-EGFP plasmid DNA (Clontech, Mountain View, CA, USA) encoding enhanced green fluorescent protein. Later, Renilla luciferase in each cell extract served as an internal control for transfection.
Expression of SP-A, SP-A-mutant and TLR4 proteins in HEK293 cells transfected with plasmid DNA constructs
The HEK293 cells transfected with pSPA or pSPA-mutant and pTLR4 constructs expressed SP-A and TLR4 proteins (Figure 1A). As expected, the molecular mass of the SP-A-mutant protein expressed by HEK293 cells transfected with pSPA-mutant construct was smaller than the full length SP-A protein expressed by HEK293 cells transfected with pSPA construct (Figure 1A). The secreted levels of SP-A were also measured in cell-free supernatants by ELISA; we measured 0.2 and 0.4 ng amounts of SP-A per µg total cellular protein in the supernatants of HEK293 cells transfected with pSPA and pSPA-mutant constructs, respectively (Figure 1A).
TLR4 was expressed on the cell surface of the pSPA or pSPA-mutant and pTLR4 co-transfected HEK293 cells, as observed by immunocytochemistry and flow cytometry (Figure 1A); intracellular expression of TLR4 was also observed.
SPA4 peptide region is important for SP-A–TLR4-interaction and inhibition of NF-κB activity
Consistent with the results published previously, 12 we observed that the SP-A interacts with TLR4 in HEK293 cells. Confocal imaging revealed the co-localization of SP-A and TLR4 in the cytoplasm, as well as at the cell-surface (Figure 1A). Importantly, the RLU values were reduced in cells co-transfected with pSPA-mutant and pTLR4 constructs compared with the cells co-transfected with pSPA and pTLR4 constructs (Figure 1B). The data suggest that the loss of SPA4 peptide region results into the reduction in SP-A–TLR4 interaction.
We extended the experiments further to assess the effects of SP-A- or SP-A-mutant TLR4 interaction on the LPS-induced NF-κB activity. The pGL4.32 NF-κB-reporter plasmid DNA was added to the two-hybrid assay system instead of pG5Luc plasmid DNA; the rest of the assay conditions were kept same. The cells were then incubated with LPS for 5 h. As anticipated, the HEK293 cells transfected with pTLR4 plasmid construct showed a significant increase in NF-κB activity against LPS-stimuli (88 RLU; Figure 1C). These results suggest that the LPS binds with TLR4 protein expressed by pTLR4-transfected cells and induces intracellular signaling. This increase in NF-κB activity was suppressed significantly in cells co-transfected with pSPA and pTLR4 constructs (88 versus 35 RLU; P < 0.001). The cells co-transfected with pSPA-mutant and pTLR4 also showed suppressed NF-κB activity (88 vs 44 RLU; P < 0.001). Although not significantly different, the NF-κB activity level was slightly increased in cells transfected with pSPA-mutant than in cells transfected with pSPA. These results suggest that reduction of interaction results in only partial inhibition of NF-κB activity.
Altogether, the results of the two-hybrid assay demonstrate that the SPA4 peptide region contributes to the SP-A-TLR4 interaction. The two-hybrid assay results with SP-A-mutant protein and TLR4 presented here confirm the findings of in silico protein–protein docking and peptide-screening analyses published earlier. 12
Physico-chemical characteristics of SPA4 peptide
Based on our previous results
12
and results from the two-hybrid assay presented here, we studied the biological effects of synthetic SPA4 peptide. The SPA4 peptide was synthesized by a commercial vendor (Genscript). Mass spectrograms and HPLC confirmed the purity of each batch of synthetic SPA4 peptide (data not shown). The SPA4 peptide is predicted to have an isoelectric point of 4.27, a net charge of −1 and an extinction coefficient of 9970 M−1* cm−1 (Figure 2A). The predicted 3D structure of SPA4 peptide exhibits beta strands and coils (Figure 2B). A plot of hydropathy index using the constants of Kyte and Doolittle indicates that the peptide is hydrophilic in nature (Figure 2C).
(A) Primary chemical structure of SPA4 peptide as predicted by PepDraw Program (Tulane University). Automated generated isoelectric point, net charge and extinction coefficient of SPA4 peptide are shown within the figure. (B) In silico predictions of 3D structure of SPA4 peptide by the PEP-FOLD online server. (C) The Kyte and Doolittle hydropathy index shows negative values indicating the hydrophilic nature of the SPA4 peptide.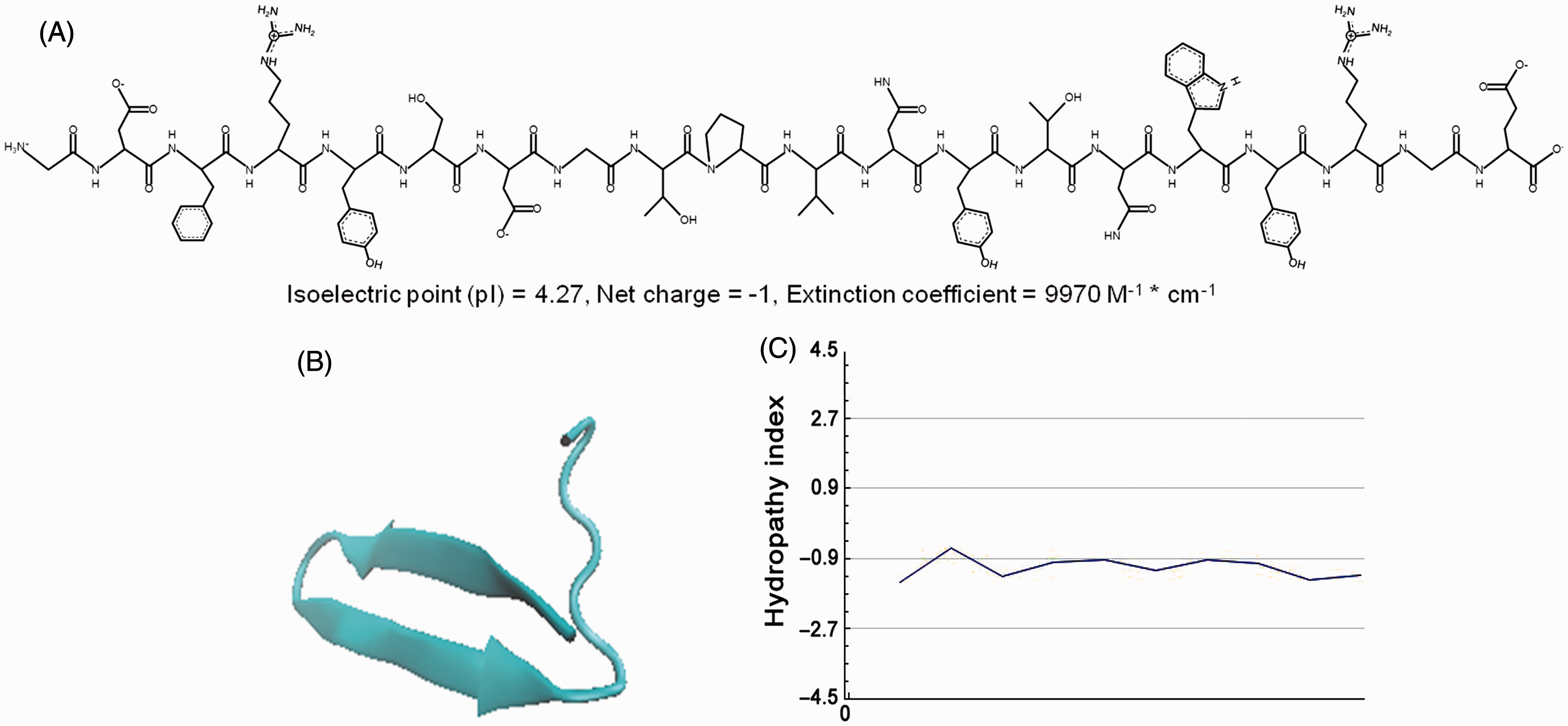
As endotoxin is a well-characterized ligand for TLR4, the presence of endotoxin can significantly influence the results. Thus, we prepared all the solutions and reagents in endotoxin-free water and performed all the assays in an aseptic environment. The endotoxin was not detectable in purified SP-A preparation, and was < 0.04 pg per µg in SPA4 peptide suspensions.
SPA4 peptide inhibits LPS-induced NF-κB activity and TNF-α
Anti-inflammatory activity of SPA4 peptide was studied in an established JAWS II dendritic cell system. The NF-κB is a transcription factor induced by LPS-TLR4 via MYD88 and TIR-domain-containing adaptor-inducing IFN-β (TRIF)-dependent pathways.29,30 The activation of NF-κB, in turn, stimulates synthesis and secretion of pro-inflammatory cytokines. The effect of SPA4 peptide on LPS-induced NF-κB was determined by investigating the expression of phosphorylated-NF-κB-p65 and NF-κB-reporter activity in a dendritic cell line. Our results show that treatment with SPA4 peptide inhibits the LPS-stimulated phospho-NF-κB-p65 expression in JAWS II dendritic cells (Figure 3A). Our results further revealed that the SPA4 peptide (1 and 10 µM) treatment significantly inhibits the LPS-induced MYD88-dependent NF-κB activity (Figure 3B) and TNF-α release (Figure 3C) in dendritic cells without any effect on the MYD88-independent NF-κB activity or the secreted levels of TNF-α. These results further support the inhibition of TLR4-induced inflammatory response by SPA4 peptide.
Effect of post-LPS treatment with synthetic SPA4 peptide on phospho-NF-κB-p65 expression, NF-κB activity and TNF-α release in JAWS II dendritic cells. (A) The JAWS II dendritic cells were challenged with LPS (100 ng/ml) for 4 h and treated subsequently with SPA4 peptide (1 μM and 10 μM) for 1 h. Twenty micrograms of total cell lysate protein was separated on 4–20% tris-glycine SDS-PAGE gel under complete reducing condition (heating + 11 mM β-mercaptoethanol) and immunoblotted with phospho-NF-κB-p65 Ab. Phospho-NF-κB-p65 and β-actin Abs-reactive bands in lysate proteins of cells treated with (lane 1) vehicle control; (lane 2) 1 µM SPA4 peptide; (lane 3) 10 µM SPA4 peptide; (lane 4:) 100 ng/ml LPS; (lane 5) 100 ng/ml LPS + 1 µM SPA4 peptide; and (lane 6) 100 ng/ml LPS + 10 µM SPA4 peptide. The bar chart in the bottom panel demonstrates the densitometric ratio of phospho-NF-κB-p65 and β-actin Ab-reactive bands. The results are from one representative experiment. Similar experiments were performed twice, separately. (B) The JAWS II dendritic cells were co-transfected with NF-κB-luciferase reporter and MYD88-dominant negative (MYD88DN) or pcDNA3.0 vector plasmid DNA constructs. Cells were stimulated with LPS (100 ng/ml) for 4 h and treated subsequently with SPA4 peptide (1 and 10 µM) for 1 h. The cells were lysed and NF-κB-associated luciferase activity was measured. Luminescence units were normalized with total cellular protein. Percent NF-κB luciferase reporter activity was compared with that in LPS-stimulated cells; P < 0.01 or P < 0.05 were noted (ANOVA); ns: not significant. (C) The TNF-α levels were measured in cell-free supernatants by ELISA and normalized with total cellular protein. P < 0.05 compared with TNF-α levels in cell-free-supernatants of LPS-treated cells (ANOVA); ns: not significant. The bars represent mean + SEM values obtained from three experiments performed in triplicate at different times.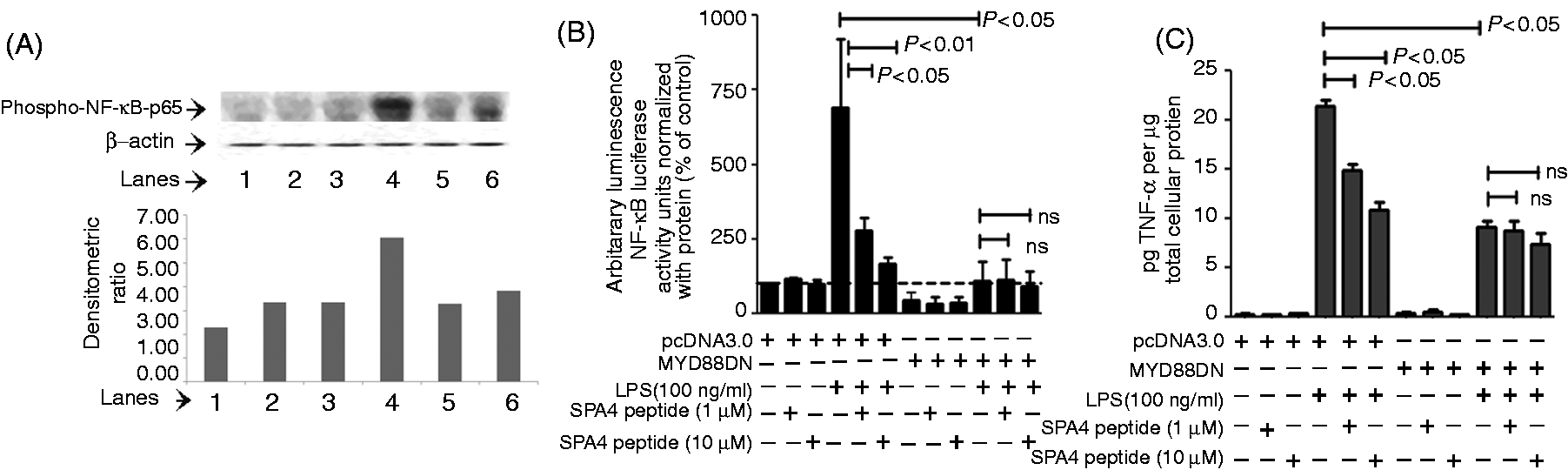
Biological effects of SPA4 peptide in a mouse model of LPS-induced lung inflammation
The activity of SPA4 peptide was then assessed by studying inflammatory parameters (nuclear localization of NF-κB-p65, TNF-α, influx of leukocytes) and endotoxic shock-like symptom indices in a mouse model of LPS-induced lung inflammation. Results were compared with those observed in SP-A-treated mice.
Inhibition of systemic TNF-α levels by SPA4 peptide translates to improvement in endotoxic shock-like symptoms
The circulating levels of LPS-induced TNF-α in mouse serum were significantly reduced after SPA4 peptide and SP-A treatment (Figure 4A). The inhibitory effect of SPA4 peptide on TNF-α was more pronounced than that of SP-A (P < 0.01 versus P = 0.09).
Effect of SPA4 peptide on LPS-induced circulating levels of (A) TNF-α and (B) endotoxic shock-like symptoms in a mouse model. Mice were challenged with LPS (15 µg per g body mass) at 0 h, treated with SPA4 peptide (2.5 µg per g body mass) or purified lung SP-A (0.5 µg per g body mass) at 1 h and sacrificed at 6 h post-LPS challenge. (A) TNF-α levels (pg/ml) were measured in serum samples of mice by ELISA. Results are shown as mean + SEM. (B) The endotoxic shock-like symptoms (ruffled fur, prostration, reactivity, diarrhea and eye exudate) were noted for each mouse on the scale of 0–3 and given an average symptom index. Results are from 6 mice in control group and 10 mice each per LPS-challenged and SP-A or SPA4 peptide treatment groups included in two separate experiments. Statistical significance was calculated by employing t-test.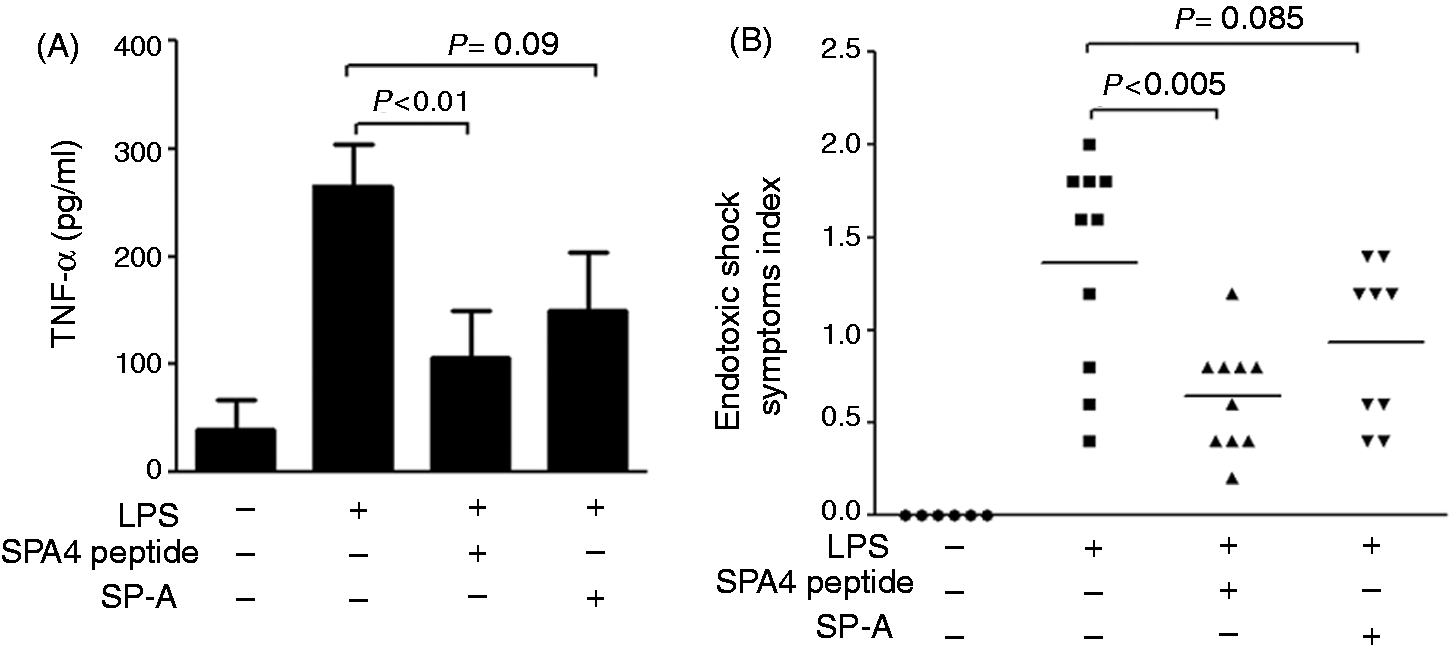
An evaluation of the endotoxic shock-like symptom indices in animals revealed an alleviation of symptoms after treatment with SPA4 peptide and SP-A. Endotoxic shock-like symptom index for each animal is demonstrated in Figure 4B. IP challenge with LPS stimulated typical symptoms of endotoxic shock (mean symptom index: 1.4) evident by ruffled fur (hair-raised and heterogenous), lack of reactivity and prostration (not reactive, difficulty in sitting and rear legs tending to be extended), diarrhea (fluidy fecal matter stuck on fur) and eye exudate (exudates and eye closed). Treatment with SPA4 peptide and SP-A led to a decrease in the LPS-stimulated endotoxic shock symptoms index (mean score 0.64 for SPA4-treated animals, P < 0.005; mean score 0.93 for SP-A treated animals, P = 0.085).
SPA4 peptide suppresses the LPS-induced TNF-α, nuclear localization of NF-κB-p65 and leukocyte influx in lung
As expected, significantly increased levels of TNF-α were noted in lung homogenates of LPS-challenged mice (P < 0.05; Figure 5). The SPA4 peptide and SP-A treatment suppressed the LPS-induced TNF-α in lung homogenates (P < 0.001 and P < 0.008; Figure 5). No significant differences were observed in MPO levels in lung tissue homogenates of LPS-challenged mice or in the LPS-challenged mice treated with SPA4 peptide or SP-A (data not shown).
Effect of SPA4 peptide on LPS-induced lung TNF-α levels in a mouse model. Mice were challenged with LPS (15 µg per g body mass) at 0 h, treated with SPA4 peptide (2.5 µg per g body mass) or purified lung SP-A (0.5 µg per g body mass) at 1 h and sacrificed at 6 h post-LPS challenge. Harvested lung tissue specimens were homogenized, and TNF-α (in pg/ml) and total protein (in µg/ml) concentrations were measured in lung tissue homogenates by ELISA and a BCA protein assay, respectively. The TNF-α levels were normalized with total lung protein amounts. The lines represent mean of results obtained from two experiments. Statistical significance was calculated by employing t-test.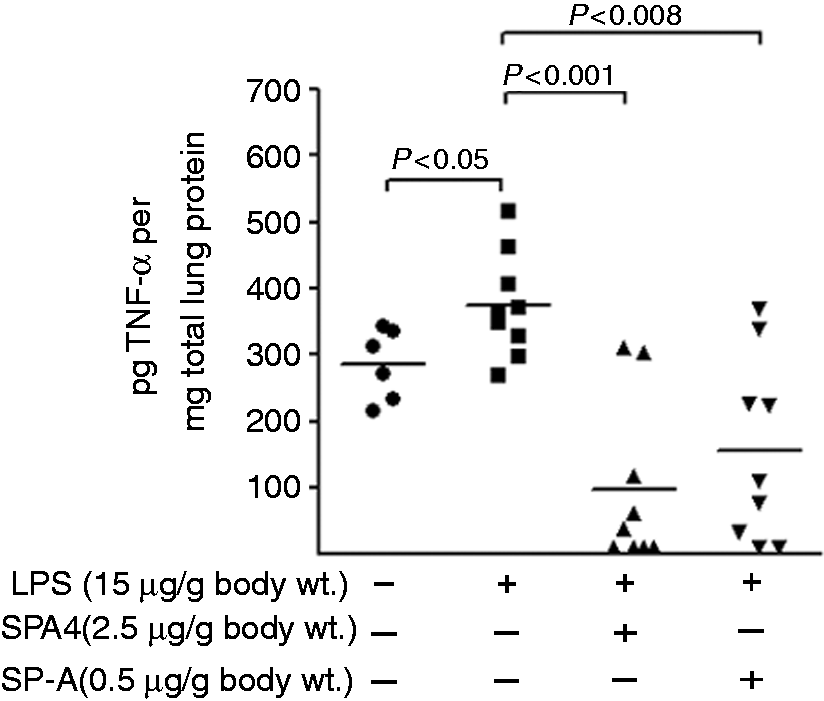
The H&E-stained lung sections were examined by a single-blinded, board-certified veterinary pathologist for the maximum lung damage present in each animal within the group. No, or minimal, damage was also reported. Firstly, LPS-induced histological changes in lung were noted primarily in the form of an accumulation of neutrophilic leukocytes within the lumen of the pulmonary vessel and/or pavemented along the endothelial lining (Figure 6A). Secondly, the leukocytes present within the lumen of the pulmonary vessels were counted. In the LPS-challenged animals, we observed more leukocytes within the central part of the lumen and pavemented along the endothelial lining. The average number of leukocytes per vessel was set at 100% in LPS-challenged mice. Percent reduction in average number of cells was calculated for SPA4 peptide- and SP-A-treated animal groups and compared with LPS-challenged mice. Upon comparison, the lungs of SPA4 peptide-treated mice revealed a 50% reduction in the number of leukocytes per vessel. SP-A treatment only resulted into 25% reduction of cell influx (Figure 6B).
SPA4 peptide treatment ameliorates the LPS-induced histological changes in lung. Mice were challenged with LPS (15 µg per g body mass) at 0 h, treated with SPA4 peptide (2.5 µg per g body mass) or purified lung SP-A (0.5 µg per g body mass) at 1 h and sacrificed at 6 h post-LPS challenge. (A) Histological observations in lung tissue sections of untreated control mice: an occasional neutrophilic leukocyte (shown as arrow) was present within the pulmonary vessels. There was no evidence of significant neutrophilic leukocyte pavementing along the endothelial lining (400×, H&E stain); LPS-challenged mice: numerous neutrophilic leukocytes were present within the lumen and observed pavementing (shown as arrows) along the endothelial lining of the pulmonary vessels (200×, 400× and 600×, H&E stain); LPS-challenged, SP-A-treated mice: numerous neutrophilic leukocytes were present within both the lumen (shown as arrows) and pavementing along the endothelial lining of the pulmonary vessel (400× and 600×, H&E stain); LPS-challenged, SPA4 peptide-treated mice: only an occasional neutrophilic leukocyte was observed within the central lumen of the pulmonary vessel. There was only minimal evidence of neutophilic leukocyte pavementing (shown as arrow) along the endothelial lining. (400× and 600×, H&E stain). (B) Number of leukocytes counted per pulmonary vessels in mice groups compared with the ones in LPS-challenged mice. The number of leukocytes per vessel in LPS-challenged mice group was set at 100% and relative percentages were calculated in other groups. Statistical significance was calculated by employing t-test.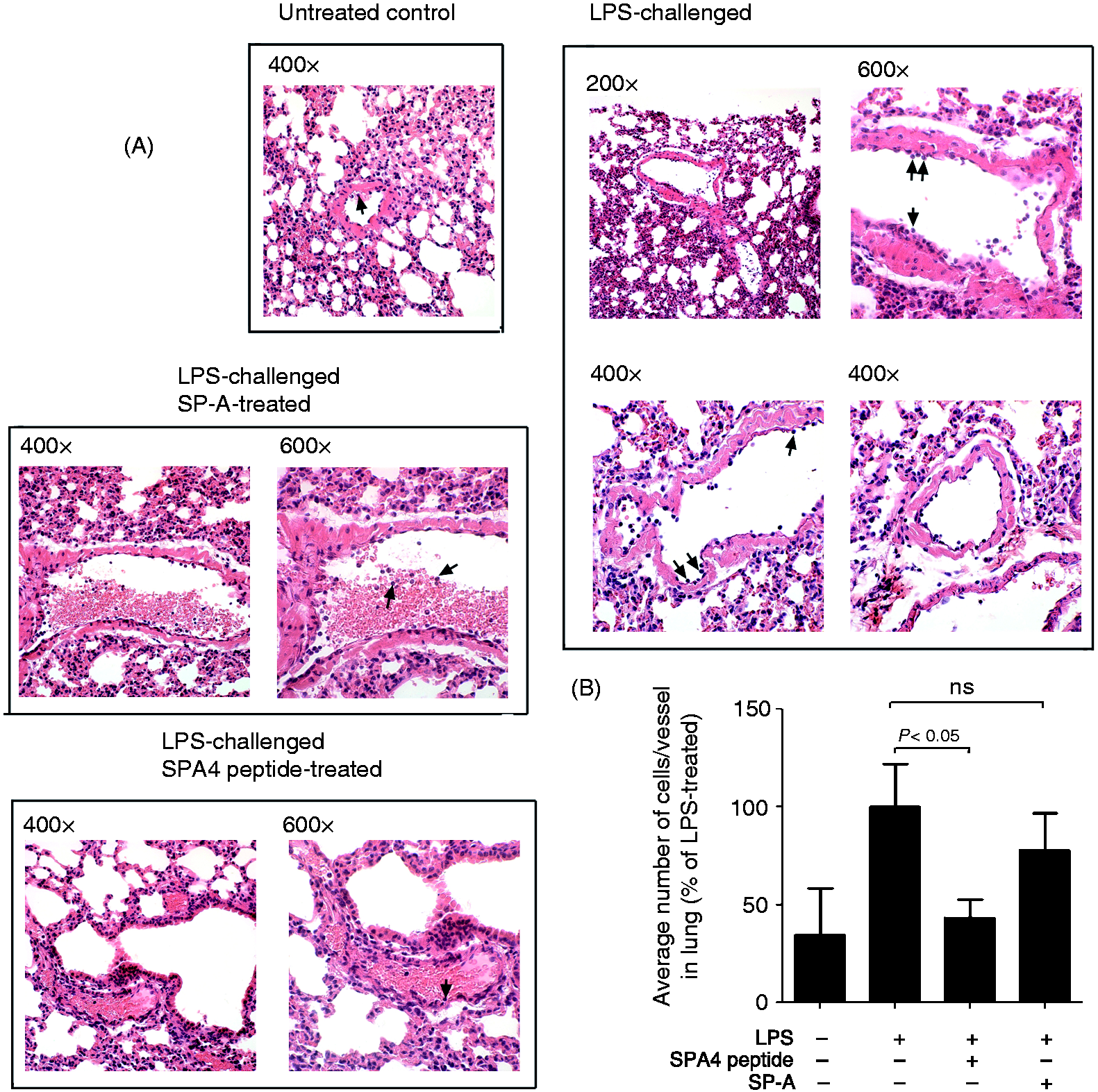
An IP challenge with LPS stimulated the nuclear localization of NF-κB-p65 in mouse lung cells. We observed that the LPS-induced nuclear staining of NF-κB-p65 was significantly reduced after treatment with SPA4 peptide and SP-A (Figure 7). However, the decrease in nuclear staining of NF-κB-p65 was more conspicuous in SPA4 peptide-treated mice than in the SP-A-treated mice. The suppression of LPS-induced nuclear localization of NF-κB-p65 in lung cells was in agreeement with the inhibitory activity of synthetic SPA4 peptide and purified lung SP-A on other inflammatory parameters.
Immunohistochemistry for endogenous expression and nuclear localization of NF-κB-p65 in lung tissues of untreated control, LPS-challenged ± SPA4 peptide- or purified lung SP-A-treated mice. Formalin-fixed lung tissue specimens were sectioned into 5 -µm sections and stained with an Ab to NF-κB-p65 (shown in blue). Finally, tissue sections were counterstained with nuclear fast red stain (shown in red). The photomicrographs were taken using 20× (upper panel) and 100× (bottom panel) objective lenses. In the bottom panel, the representative area within each photomicrograph is enlarged for better visualization. The arrows within the photomicrograph indicate nuclear localization of NF-κB-p65.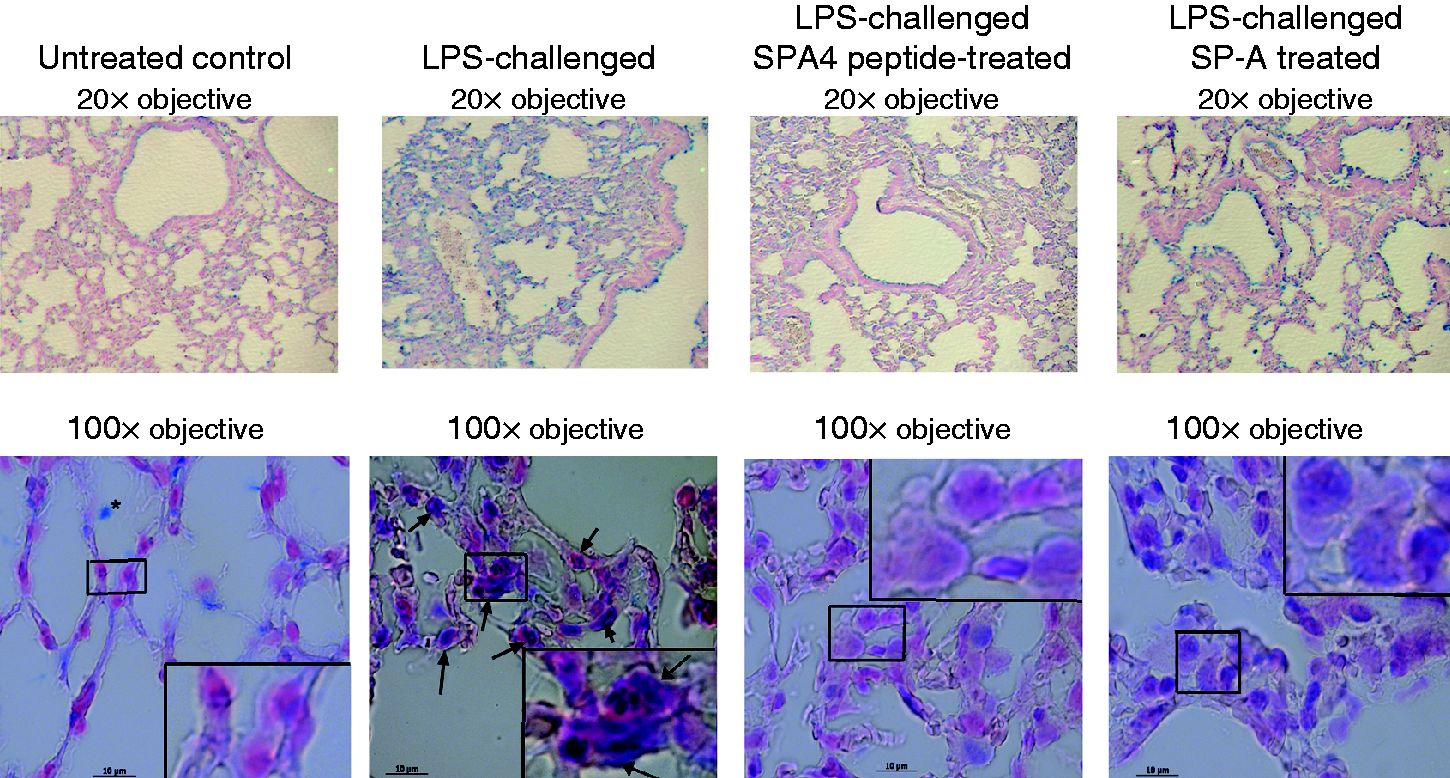
Discussion
SP-A plays an important role in host defense against a variety of pathogenic insults; 7 SP-A induces phagocytosis of bacterial and fungal pathogens, and suppresses the inflammatory response.31–35 As per the published results in animal models and patients, a decrease in the amounts of SP-A in bronchoalveolar lavage fluids is associated with fulminant lung infection and inflammation;36–40 thus, the utilization of SP-A as a therapeutic has been of a contemporary interest. In the past, it has not been possible to develop an SP-A-based therapeutic or an SP-A-containing clinical surfactant because of the large size and hydrophilicity of SP-A. In general, large-sized proteins tend to induce a non-specific immune response and are cleared rapidly; its hydrophilic nature also makes it difficult to mix SP-A with hydrophobic lipids of clinical surfactants. An interesting study from Gardai et al., 41 demonstrated that the N-terminal region of SP-A can also induce pro-inflammatory effects against infectious challenge through its interaction with calreticulin. In view of these published results and formulation-related issues with full-length SP-A, we propose that the small SP-A fragments mimicking the beneficial host defense characteristics of SP-A can be of therapeutic use.
In a recently published article, we reported that the SP-A interacts with TLR4, and SP-A–TLR4 interaction suppresses the inflammatory response but maintains the phagocytic uptake of bacteria. 13 These results corroborated with published reports in the literature (reviewed in Pastva et al. 7 and Awasthi 42 ) Subsequently, we developed a unique approach to identify SP-A regions that can mimic at least some of these host defense properties of SP-A through their interaction with TLR4. We obtained an in silico computer model of SP-A–TLR4–MD2 complex, synthesized a small library of SP-A peptide fragments derived from the interface of SP-A–TLR4 complex and screened for an inhibition of LPS-induced TNF-α in a dendritic cell line. 12 One of the peptides, SPA4 derived from the C-terminal region of SP-A, was identified to suppress the release of TNF-α against LPS stimulus. 12 We had utilized the in silico modeling and in vitro binding assay; biological relevance of SP-A–TLR4 interaction or the SPA4 peptide region was not studied. 12 Thus, herein we investigated the contribution of SPA4 peptide region in SP-A–TLR4 interaction in HEK293 cells using a two-hybrid assay and evaluated the biological effects of synthetic SPA4 peptide in a dendritic cell system and in a mouse model of inflammation induced by TLR4 ligand-LPS. Presumably, the IP LPS challenge model in mice would closely mimic the pathological scenario, as seen in patients with endotoxic shock-induced ARDS. 43 We anticipate that TLR4-signaling would be activated. Thus, the introduction of SPA4 peptide may help improve the host defense and alleviate the clinical symptoms.
To understand the activity of SPA4 peptide at cellular level we included HEK293 cells and JAWS II dendritic cells. The HEK293 cells provided a cleaner system to test the SP-A-TLR4 interaction without any interference from endogenous SP-A and TLR4. To our knowledge, the standard methods (co-localization, co-immunoprecipitation–immunoblotting) commonly employed for studying protein–protein interaction have a limited capacity of quantitating the affinity or avidity, and identifying the regions involved in the protein–protein interaction. Thus, the two-hybrid assay in HEK293 cells could provide a better alternative to identify interacting domains and regions. Our results presented herein indicate that the two-hybrid assay provides quantitative measurement of protein–protein interaction and functional relevance, and allows investigation of particular domains and regions. The type II lung epithelial cells possess a highly organized system for post-translational modification, packaging (e.g. lamellar body structures) and secretion of SP-A.44,45 Although HEK293 cells may not have this machinery, we detected a measurable quantity of secreted SP-A in the supernatants of pSPA and pSPA-mutant-transfected HEK293 cells (Figure 1A). The work presented herein in the HEK293 cell system establishes that the SPA4 peptide region is important for interaction with TLR4 and inhibition of LPS–TLR4-induced NF-κB. Overall, our results suggest that the two-hybrid assay in HEK293 cells can provide a useful high-throughput tool for identifying other regions of SP-A that bind to TLR4 and modulate immune responses.
As the HEK293 cells are not derived from peripheral mucosal sites and may not mimic the natural scenario, we assessed the biological activity of synthetic SPA4 peptide in vitro in a murine bone marrow-derived dendritic cell system and in vivo in a mouse model of LPS-induced lung inflammation.
The results we obtained in mouse dendritic cells reveal that the SPA4 peptide inhibits the LPS-stimulated phosphorylation of NF-κB-p65 unit, NF-κB activity and TNF-α release in a MYD88-dependent manner (Figure 3). We also found that the SPA4 peptide did not bind to LPS. 46 Overall, these results strengthen our broad hypothesis that the activity of SPA4 peptide against LPS stimuli (TLR4 ligand) is most likely through its interaction with TLR4 and not by sequestering its ligand—LPS. Detailed studies are required to delineate the mechanism of action of SPA4 peptide and other TLR4-interacting regions of SP-A.
In a very recent study, we utilized an established human colonic cancer epithelial cell line that constitutively expresses TLR4; 46 the biological effects of SPA4 peptide in other TLR4-expressing cells and animal models remain to be explored. In this very initial study, we included a mouse model of LPS-induced lung inflammation. A single time point included in this study may not be optimum for an assessment of significant changes in MPO; further comprehensive investigations are now being undertaken. Future studies in genetically-modified mouse models will help us understand the mechanism of action of SP-A and SP-A-derived regions through their interaction with TLR4. Our results reveal that the SPA4 peptide suppresses the LPS-induced inflammatory parameters (TNF-α, NF-κB activity and leukocyte influx), and alleviates LPS-induced symptoms. Interestingly, the anti-inflammatory effects of SPA4 peptide were equal to or more pronounced when compared with full-length SP-A (Figures 4–7). Several possibilities exist, including the specific targeting of TLR4 by SPA4 peptide for an inhibition of inflammation. Full-length SP-A, however, can exert both pro-inflammatory and anti-inflammatory effects through a number of cell receptors and mechanisms.47–49 The mechanism of action of SPA4 peptide may differ from that of full-length SP-A. Additional studies are required to address these aspects.
We believe that the results of this study can be of clinical importance because an overwhelming inflammation leads to ARDS and multiple organ failure,3,50 and an increased expression and activity of TLR4 has been linked with deleterious inflammatory response.51–54 Thus, we envision that the TLR4-interacting SPA4 peptide and other regions of SP-A may have therapeutic potential in ARDS. Moreover, none of the clinical surfactants have SP-A or SP-D. 55 A comprehensive evaluation of the SPA4 peptide and other TLR4-interacting regions of SP-A may facilitate the design of a novel SP-A-based immunomodulator or clinical surfactant.
Footnotes
Parts of this work were presented at the 2011 OUHSC-Summer undergraduate research program, 2011 OUHSC-Graduate Research, Education and Technology Symposium and 2012 Gordon Research Conference on the “Biology of Acute Respiratory Infection”.
Acknowledgements
We thank Julie Maier, Imaging Core Facility, Oklahoma Medical Research Foundation, Oklahoma City for providing expertise in performing immunohistochemistry; and Jim Henthorn, Flow and Imaging Core Facility, OUHSC, Oklahoma City for providing help with confocal microscopy. Finally, contributions from Rebecah Ritchie, a summer undergraduate student (between May and July 2011) in SA’s laboratory supported by NIH-INBRE to OUHSC are also appreciated.
Funding
This work was supported by funding from American Heart Association Grant-in-Aid.