
Abstract
It is now established that IL-17 has a broad pro-inflammatory potential in mammalian host defense, in inflammatory disease and in autoimmunity, whereas little is known about its anti-inflammatory potential and inhibitory feedback mechanisms. Here, we examined whether IL-17A can inhibit the extracellular release of IL-23 protein, the upstream regulator of IL-17A producing lymphocyte subsets, that is released from macrophages during pulmonary inflammation. We characterized the effect of IL-17A on IL-23 release in several models of pulmonary inflammation, evaluated the presence of IL-17 receptor A (RA) and C (RC) on human alveolar macrophages and assessed the role of the Rho family GTPase Rac1 as a mediator of the effect of IL-17A on the release of IL-23 protein. In a model of sepsis-induced pneumonia, intravenous exposure to Staphylococcus aureus caused higher IL-23 protein concentrations in cell-free bronchoalveolar lavage (BAL) samples from IL-17A knockout (KO) mice, compared with wild type (WT) control mice. In a model of Gram-negative airway infection, pre-treatment with a neutralizing anti-IL-17A Ab and subsequent intranasal (i.n.) exposure to LPS caused higher IL-23 and IL-17A protein concentrations in BAL samples compared with mice exposed to LPS, but pre-treated with an isotype control Ab. Moreover, i.n. exposure with IL-17A protein per se decreased IL- 23 protein concentrations in BAL samples. We detected IL-17RA and IL-17RC on human alveolar macrophages, and found that in vitro stimulation of these cells with IL-17A protein, after exposure to LPS, decreased IL-23 protein in conditioned medium, but not IL-23 p19 or p40 mRNA. This study indicates that IL-17A can partially inhibit the release of IL-23 protein during pulmonary inflammation, presumably by stimulating the here demonstrated receptor units IL-17RA and IL-17RC on alveolar macrophages. Hypothetically, the demonstrated mechanism may serve as negative feedback to protect from excessive IL-17A signaling and to control antibacterial host defense once it is activated.
Keywords
Introduction
IL-17A (synonymous to IL-17) is a cytokine that is now emerging as critical for host defense against bacteria, virus and fungi.1–5 Many of its properties relate to local actions at the site of infection, including the recruitment of neutrophils, and the induction of extracellular and intracellular microbial killing in phagocytes, as well as the release of anti-bacterial mediators from structural cells.1,5–11 Several of these actions promote inflammation and, consequently, IL-17A has mainly been regarded as a cytokine with pro-inflammatory actions. 12
Interestingly, there is now an accumulating body of evidence for IL-17A being capable of exerting anti-inflammatory actions as well, depending upon setting and timing. Thus, the inhibition of endogenous IL-17A actually worsens inflammation at certain time points in several settings, including animal models of airway allergy, inflammatory bowel disease and atherosclerosis.13–17 In line with these latter findings, stimulation with recombinant IL-17A protein exerts anti-inflammatory effects at certain time points in animal models of gastritis, experimental autoimmune neuritis and chronic relapsing uveitis.18–20 In further support of IL-17A being capable of exerting anti-inflammatory actions, the stimulation with recombinant IL-17A protein increases neutrophil apoptosis, as well as the phagocytosis of apoptotic neutrophils by macrophages in vitro, suggesting a role in resolving inflammation, as well in initiating it. 21
IL-23 is a member of the IL-12 family of cytokines, and it is believed to be an important regulator of the expansion and stabilization of the IL-17A-producing phenotype of lymphocytes, such as T helper cell (Th) 17, γδT and NK T cells.22–28 This cytokine is thereby not only promoting the production of IL-17A, but also that of IL-17F and IL-22, cytokines that may be produced by the very same lymphocyte subsets as IL-17A and all of which are important mediators of host defense in several organs, including the lungs.24,26,29,30 Notably, IL-23 is produced by cells of the monocyte lineage in response to stimulation pattern recognition receptors.31–33 Moreover, it has recently been demonstrated that IL-25, the IL-17A-related cytokine formerly named IL-17E, actually decreases the production of IL-23.31–33 This results in a subsequent decrease in the release of IL-17A from mouse macrophages and dendritic cells from large intestine, as well as human CD14+ cells from the gut of patients with Crohn’s disease.31–33 Of particular interest, just like IL-17A, IL-25 acts via the IL-17 receptor A (RA) subunit and it signals through the intracellular adaptor molecule Act1.34,35 Given this, and the abundance of alveolar macrophages present in the airways, we hypothesized that stimulation with IL-17A decreases the release of IL-23 protein to the extracellular space at certain time points during pulmonary inflammation. 36
The main aim of the current study was to determine whether stimulation with IL-17A inhibits the extracellular release of IL-23 protein, the upstream regulator of IL-17A-producing lymphocyte subsets, during pulmonary inflammation. To do a functional evaluation of this, we applied a broad methodological approach in vivo, as well as in vitro. We used three different experimental models in vivo: first, we used a model of sepsis-induced pneumonia where wild type (WT) and IL-17A knockout (KO) mice were exposed to viable Staphylococcus aureus intravenously; second, we used a model of local Gram-negative airway infection where mice were exposed to endotoxin (LPS) from Escherichia coli via administration intranasally (i.n.) with and without pre-treatment with a neutralizing anti-IL-17A Ab; third, and last, we used a model of the direct effect of IL-17A per se locally in the airways where mice were stimulated with recombinant IL-17A protein i.n. Moreover, for the evaluation in vitro, we used three types of human cells of the monocyte-lineage, including alveolar macrophages, for which recombinant IL-17A protein was applied as a stimulus after exposure to LPS. We also analyzed the expression of both IL-17RA and IL-17RC on human alveolar macrophages in vitro, as this expression is likely to be an important prerequisite for IL-17A’s specific actions on these cells. In addition, we determined the effect of IL-17A on mRNA levels for the IL-23 subunits p19 and p40, respectively, and we evaluated the involvement of Rac1, a Rho family GTPase, in mediating the effect of IL-17A on the extracellular release of IL-23 protein caused by exposure to LPS in vitro.37,38
Material and methods
Ethics statement
Permission for studies on mice was obtained from the ethical Committee for Animal Studies in Gothenburg, Sweden (diary numbers: 138-2005, 293-2008 and 106-2009), in accordance with national animal welfare legislation. The study protocol for bronchoalveolar lavage on humans was approved by the Ethics Committee in Gothenburg
Mice
Male BALB/c mice (8–11 wk old; Taconic, Ejby, Denmark), C57BL/6 WT mice (10–15 wk old; Scanbur, Sollentuna, Sweden) and C57BL/6 IL-17 KO mice (10–15 wk old) were kept under standard conditions in a 12 h day–night rhythm with access to food and water ad libitum. 39
Administration of S. aureus, LPS and recombinant cytokine in mice
In the model of sepsis-induced pneumonia, IL-17A KO and WT C57BL/6 mice were exposed systemically by inoculating them i.v. in the tail vein with 0.22 × 108 bacteria/mouse of toxic shock syndrome toxin-1-producing S. aureus LS-1 in a total volume of 200 µl PBS.40,41 Viable counts were performed to determine the number of bacteria injected. Twenty-four h after inoculation of S. aureus, IL-17A KO and WT mice were anesthetized by i.p. injection of ketamine (75 mg/kg; Ketalar; Pfizer, Sollentuna, Sweden) and medetomidine (1 mg/kg; Dormitor Orion pharma, Espoo, Finland) and euthanized by bleeding of the aorta, followed by cervical dislocation.
In the models of Gram-negative airway infection and local effect of IL-17A per se, BALB/c mice were anaesthetized transiently using isofluorane (Apoteksbolaget, Gothenburg, Sweden). After this, the mice were either exposed to LPS (10 µg, E. coli serotype 026:B6; Sigma-Aldrich, St. Louis, MO, USA) or stimulated by recombinant mouse (rm) IL-17A protein [(3 µg) Cat. no. 421-ML; R&D Systems, Minneapolis, USA] or corresponding vehicle in 50 µl of PBS through local administration i.n.. It is known from our previous studies that the utilized dose of IL-17A induces reproducible effects in the airways of mice in vivo. 42 A subgroup of mice were pre-treated with i.p. injection of 200 µg neutralizing monoclonal anti-mouse IL-17A Ab (aIL-17 Ab) (Cat. no. MAB 421; R&D Systems) or the isotype control, IgG2a Ab (Cat. no. MAB006; R&D Systems) in 500 µl, 24 h before exposure to LPS. The mice were euthanized after 24 h (LPS-treated mice) or 2 h (rmIL-17A-treated mice) by bleeding the right ventricle of the heart after anesthesia using ketamine (670 mg/kg, Ketalar; Pfizer) and xylazine (130 mg/kg, Rompun; Bayer, Leverkusen, Germany) i.p. The short incubation time was chosen for the experiments on the effect of rmIL-17A protein to avoid confounding effects of newly recruited neutrophils and monocytes.
Bronchoalveolar lavage in mice
After establishing a tracheotomy, bronchoalveolar lavage (BAL) was performed (4 × 0.5 ml for IL-17A KO and WT C57BL/6 mice; 2 × 0.25 ml for BALB/c mice administered aIL-17A Ab or IgG2a Ab; and 4 × 1.0 ml for BALB/c mice administered rmIL-17A or its vehicle), using Hank’s Balanced Salt Solution (HBSS; Sigma-Aldrich). For each mouse, all the recovered BAL samples were pooled and kept on ice until centrifugation (10 min, 4℃ at 300 g). After centrifugation, the cell-free BAL fluid was frozen (−80℃) for later analysis. The BAL cells in the pellets of centrifuged BAL samples were re-suspended in PBS, total cell number was determined using a Bürker-chamber and cytospin slides were prepared. In order to assess the total number of macrophages and neutrophils in the BAL samples, cell differential counts were determined by counting 400 cells per cytospin slide stained in May-Grünewald-Giemsa staining, as described elsewhere. 8
Culture of bacteria from lung tissue of mice
Both lungs from IL-17A KO and WT C57BL/6 mice that had been exposed systemically to S. aureus were dissected aseptically, kept on ice, homogenized, serially diluted in PBS and spread on blood agar plates. The number of CFUs per lung pair was determined after 24 h of incubation at 37℃.
BAL for harvest of alveolar macrophages in humans
The study protocol for BAL on humans in Gothenburg has been used previously in a recently published study. 43 The investigated subjects were non-smoking and non-atopic (i.e. negative Phadiatop™ test) individuals without any regular medication who constituted technical control subjects for a larger study. 43 The subjects had normal ventilatory lung function, normal clinical status and electrocardiogram, in accordance with published study inclusion criteria. 43 Bronchoscopy with bilateral BAL was performed on each subject using 3 × 50 ml of PBS on each side. Bronchoscopy with lavage was also carried out at the Royal Liverpool and Broadgreen University Hospitals Trust, with similar and previously published 44 protocols. Volunteers recruited in Liverpool were all healthy, non-smoking adults, without any regular medication.
IL-17RA and IL-17RC on human alveolar macrophages
BAL cells collected for the flow cytometry analysis of IL-17RA and IL-17RC were placed directly onto ice following collection and processed immediately for staining of the referred cell surface receptors without an adherence step. The BAL cells (0.5–1 × 105) in RPMI were pre-incubated with irrelevant IgG to block Fc receptors on alveolar macrophages for 15 min on ice. Cells were then stained separately with equal concentrations of the following mouse anti-human mAbs, IL-17RA or IgG1 conjugated with AF647 (BioLegend, San Diego, CA, USA) or IL-17RC or IgG2b APC (R&D Systems) and incubated on ice for a further 15 min. Cells were washed once and more than 10,000 events were acquired on a BD LSR 2 (BD Biosciences, Franklin Lakes,NJ, USA). Compensation settings were set manually and identically for each Ab pair. The data set was analyzed using Flowjo software (Treestar, Ashland, OR, USA).
Culture of human cells of the monocyte lineage
Monocytes were harvested from the venous blood of healthy human volunteers. Peripheral blood mononuclear cells were collected by density centrifugation over a Ficoll gradient (Ficoll-Paque Plus; GE Healthcare, Uppsala, Sweden). Monocytes were then isolated using negative selection using Monocyte Isolation Kit II (Miltenyi Biotec, Bergisch Gladbach, Germany). To obtain monocyte-derived macrophages, monocytes were cultured at a concentration of 1 × 106/ml at 37℃ and 5% CO2 in supplemented medium (RPMI 1640 with 10% FBS, 1% penicillin-streptomycin, 1 mM sodium pyruvate and 2 mM
Analyses of gene expression by RT-PCR
RNA was prepared from lysed in vitro cultured and stimulated human monocyte-derived macrophages and BAL macrophages using RNeasy® mini kit, Qia Shredder and RNAse-free DNAse (all from Qiagen). cDNA was synthesized using High Capacity Reverse Transcription cDNA kit (Applied Biosystems, Foster City, CA, USA) with RNase inhibitor (Qiagen). One microliter of the cDNA was used for a 20-μl Taqman assay. Each sample was run in duplicate. The assay was run with Taqman® Universal PCR Mastermix on 7500 Real Time PCR System with Taqman® Gene Expression for IL-23 p19 inventoried assay Hs00372324_m1 and IL-23 p40 inventoried assay Hs01011515_m1 (both from Applied Biosystems). Human β-actin endogenous control 4333762F (Applied Biosystems) was used to normalize the data.
ELISA
The cell-free BAL fluid from mice, as well as the cell-free medium of human monocytes, monocyte-derived macrophages and alveolar macrophages cultured in vitro, was analyzed for IL-23 (p19 and p40) protein using ELISA [Cat. no. 88-7230 (detection limit: 7.8 pg/ml) and 88–7237 (detection limit: 15.6 pg/ml) respectively; eBioscience, San Diego, CA, USA). In addition, the cell-free medium of human monocytes and monocyte-derived macrophages cultured in vitro was analyzed for IL-12 (p70) protein using ELISA [Cat. no. DY1270 (detection limit: 15.6 pg/ml); R&D Systems]. Cell-free BAL fluid from mice stimulated with LPS or vehicle i.n. was analyzed for IL-17A and IL-22 protein using ELISA [Cat. no. M1700 (detection limit: 10.9 pg/ml) and D2200 (detection limit: 7.8 pg/ml) respectively; R&D Systems]. All ELISAs were performed according to the manufacturer’s instructions.
An interference test was performed to exclude interference of aIL-17A Ab or its isotype on the IL-17A ELISA. For this, samples containing recombinant IL-17A (88 and 355 pg/ml) taken from the IL-17A standard of the kit and either aIL-17A Ab (50 or 1000 ng/ml) or its isotype control IgG2a (50 or 1000 ng/ml), or calibration diluent was prepared. The standard curve of the IL-17A ELISA-kit ranged from 10.9 to 700 pg/ml. The higher concentration (1000 ng/ml) of aIL-17A Ab has been shown previously to be a sufficient concentration to neutralize 90% of the rmIL-17A activity in NIH 3T3 mouse fibroblasts when 10 ng/ml IL-17 was used. 46 The lower concentration (50 ng/ml) of aIL-17A Ab was chosen to correspond to the concentration range of IL-17A protein that was used in this test. The samples were prepared in triplicates and incubated for 30 min. After this, the samples were added to the IL-17A ELISA plate as triplicates, and then the instructions in the product manual of the ELISA-kit were followed. The interference test showed that when the low concentration (50 ng/ml) of the Abs were tested, there was no difference in the measured IL-17A protein concentration between the negative control (calibrator diluent) and the Ab-containing samples for neither anti-IL-17A Ab nor IgG2a (the concentrations of recombinant IL-17A protein added to the test were 88 and 355 pg/ml; the measured concentrations were: vehicle—89 and 359; IgG2a—92 and 378; anti-IL-17A Ab—86 and 363 pg/ml IL-17). However, in the measured IL-17A protein concentration in the samples where the higher concentration (1000 ng/ml) of aIL-17A Ab was used, there was a decrease of approximately 25% compared with the negative control. This was the case for both of the IL-17A concentrations used (the concentration of recombinant IL-17A protein added to the test were 88 and 355 pg/ml; the measured concentrations were: vehicle—89 and 359; IgG2a—105 and 364; anti-IL-17A Ab—64 and 273 pg/ml IL-17A). Thus, the presence of aIL-17A Ab or its isotype control IgG2a Ab is not affecting the concentration of mouse IL-17A protein in the BAL samples detected by the relevant ELISA kit.
For every matched set of experiments in vitro we utilized non-detectable IL-23 concentrations in the conditioned cell medium from cells exposed to LPS as an exclusion criterion. This led to the exclusion of two experiments.
Statistical analysis
Data are expressed as mean (SEM) or mean (SD) as indicated. For all experiments, each donor contributed to one independent experiment; n represents the number of independent experiments. Differences were considered to be statistically significant when P ≤ 0.05. Two-tailed Student’s t-test was used for statistical analysis of data from mouse BAL samples, human monocyte-derived macrophages and alveolar macrophages in vitro unless otherwise stated. Spearman’s rank correlation test was used to analyze the correlations between the concentration of alveolar macrophages and IL-23 concentrations from mouse BAL samples using the program IBM SPSS. The IL-23 concentrations in the cell culture medium of the human monocyte-derived macrophages and the alveolar macrophages were transformed logarithmically in order to normalize the baseline variance. The concentration–response data from Rac1-inhibitor-treated human monocyte-derived macrophages in vitro were analyzed by using repeated measures ANOVA.
Results
Local IL-23 protein in the model of sepsis-induced pneumonia for WT and IL-17A KO mice
In our model of sepsis-induced pneumonia, we administered S. aureus systemically in WT and IL-17A KO C57BL/6 mice through i.v. inoculation and subsequently measured the concentrations of IL-23 protein in cell-free BAL fluid. We found that the concentration of IL-23 protein in cell-free BAL fluid was significantly lower in WT mice than in IL-17A KO mice 24 h after, but not before, i.v. inoculation of S. aureus (Figure 1A). The concentration of alveolar macrophages in BAL samples was lower in the WT mice compared with IL-17A KO mice after, but not before, i.v. inoculation of S. aureus (Figure 1B). There was no statistically significant correlation between the concentration of BAL macrophages and the concentration of IL-23 in the BAL samples (Spearman’s rho = 0, P = 1 for uninfected IL-17A KO mice; rho = −0.6, P = 0.4 for uninfected WT mice; Spearman’s rho = 0, P = 1 for S. aureus-infected IL-17A KO mice; Spearman’s rho = −0.6, P = 0.285 for S. aureus-infected WT mice) (Supplementary Figure 1A, B). Under these conditions there was no substantial number of neutrophils in the BAL samples (<2%, data not shown). There was also an apparent tendency, although not significant, towards a lower number of CFUs in the lung tissue of WT mice compared with IL-17A KO mice (Figure 1C).
Concentrations of extracellular IL-23 protein and macrophages in BAL samples and bacterial load per lung pair from WT and IL-17A KO mice after systemic exposure to Staphylococcus aureus. IL-17A KO and WT C57BL/6 mice were inoculated i.v. with S. aureus (0.2–1 × 108 per mouse). Mice were sacrificed either before (uninfected, 0 h), or after (infected, 24 h) inoculation i.v. BAL was performed using 4 × 0.5 ml HBSS (n = 4-5). n represents the number of mice in each group. (A) Concentration of IL-23 protein in cell-free BAL fluid. (B) Concentration of alveolar macrophages in BAL samples. (C) Bacterial load; number of CFUs of S. aureus per lung pair at 24 h after infection. Results are shown as mean with SEM bars. *P < 0.05; **P < 0.01; ns: P ≥ 0.05.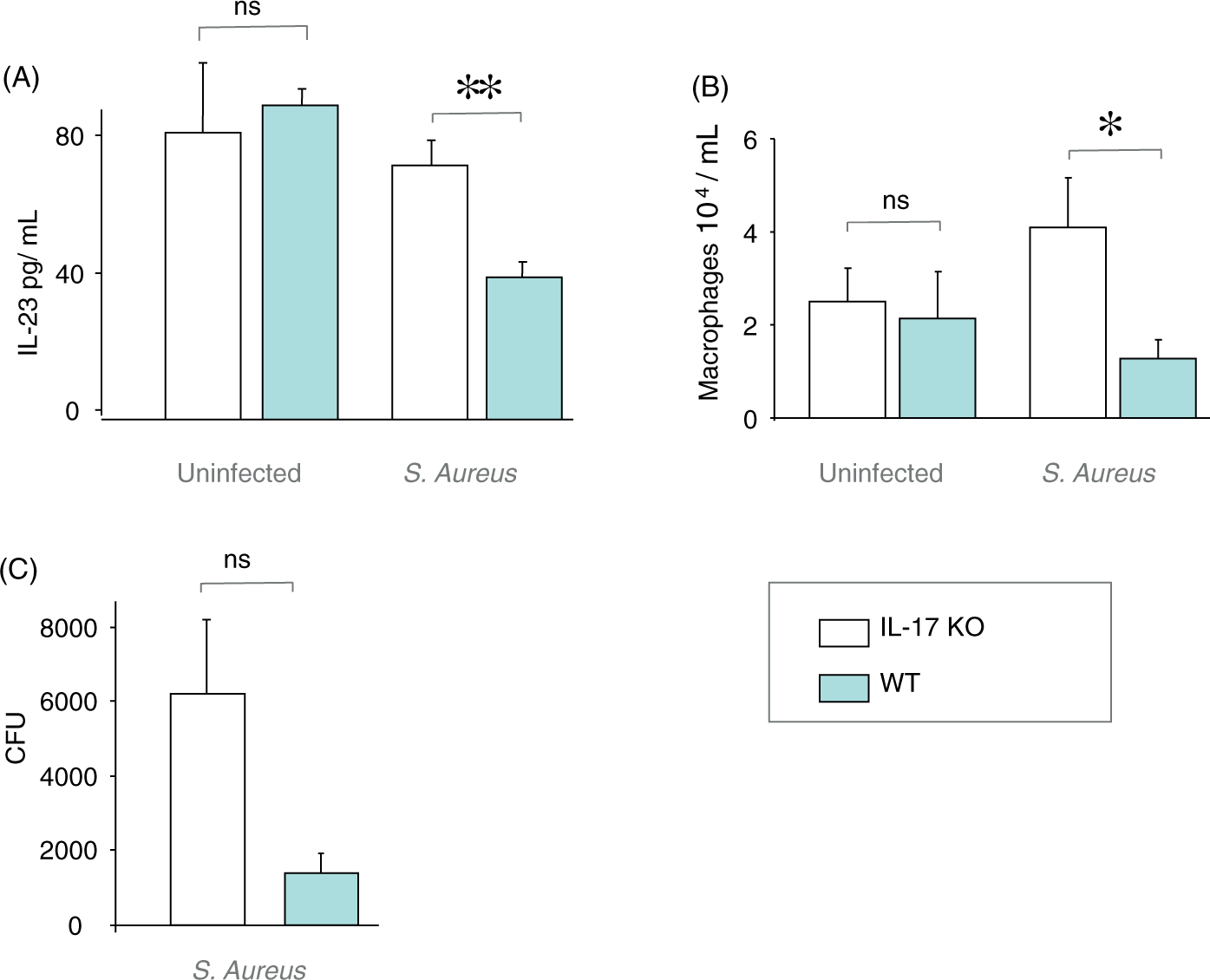
Local IL-23 protein in the model of Gram-negative airway infection, with and without pre-treatment with an anti-IL-17A Ab
In our model of Gram-negative airway infection, we exposed BALB/c mice locally to LPS through i.n. instillation 24 h prior to sampling and added a systemic intervention by using i.p. pre-treatment with aIL-17A Ab or its isotype control IgG2a. In this model, LPS exposure caused a significant decrease in the concentration of IL-23 protein in the cell-free BAL fluid from mice exposed to LPS compared with vehicle control for LPS (Figure 2A). The IL-23 concentration was significantly increased in the cell-free BAL fluid samples after exposure to LPS and pre-treatment with aIL-17A Ab compared with LPS exposure plus pre-treatment with isotype control (IgG2a) (Figure 2A). The concentration of alveolar macrophages was significantly decreased in the BAL samples after exposure to LPS and IgG2a compared with vehicle control for LPS. However, there was no reproducible difference in the concentration of alveolar macrophages when compared with LPS exposure plus pre-treatment with aIL-17A Ab (Figure 2B). There was a statistically significant correlation between the concentration of IL-23 and the concentration of BAL macrophages in the BAL samples after LPS exposure and pre-treatment with aIL-17A Ab (Spearman’s rho = −0.857, P = 0.014), but no correlation after exposure to LPS (Spearman’s rho = −0.139, P = 0.701) or vehicle control (Spearman’s rho = −0.4, P = 0.29) together with pre-treatment with IgG2a (Supplementary Figure 2). The concentration of neutrophils was increased in the BAL samples after LPS exposure plus treatment with IgG2a, both when compared with vehicle control for LPS and with LPS exposure plus pre-treatment with aIL-17A Ab (Figure 2C). There was a statistically significant increase in the concentration of both IL-17A and IL-22 protein in the cell-free BAL fluid after LPS exposure and pre-treatment with IgG2a compared with vehicle control for LPS (Figure 2D, E). The concentration of IL-22 protein, though not as substantial compared with that of IL-17A, was further increased in the corresponding samples after LPS exposure plus pre-treatment with aIL-17A Ab compared with LPS exposure plus treatment with IgG2a (Figure 2D, E). The concentrations of IL-17A protein in the samples from control mice treated with vehicle for LPS were all below the detection limit.
Extracellular concentrations of cytokines and cells in BAL samples after local exposure to endotoxin (LPS) with and without neutralization of endogenous IL-17A. BALB/c mice were pre-treated i.p. with anti-mouse IL-17A Ab (aIL-17A Ab: 200 µg) or isotype control Ab (IgG2a) and then instilled with LPS (10 µg) or vehicle intranasally over 24 h. The BAL was performed using 2 × 0.25 ml HBSS. n represents the number of mice in each group. (A) Concentration of IL-23 protein in cell-free BAL fluid (n = 8–10). (B) Concentration of alveolar macrophages in BAL samples (n = 9–10). (C) Concentration of neutrophils in BAL samples (n = 9–10). (D) Concentration of IL-17A protein in cell-free BAL fluid (n = 4–5). (E) Concentration of IL-22 protein in cell-free BAL fluid (n = 9–10). Results are shown as mean with SEM bars. *P < 0.05; **P < 0.01; ***P < 0.001; ns: P ≥ 0.05.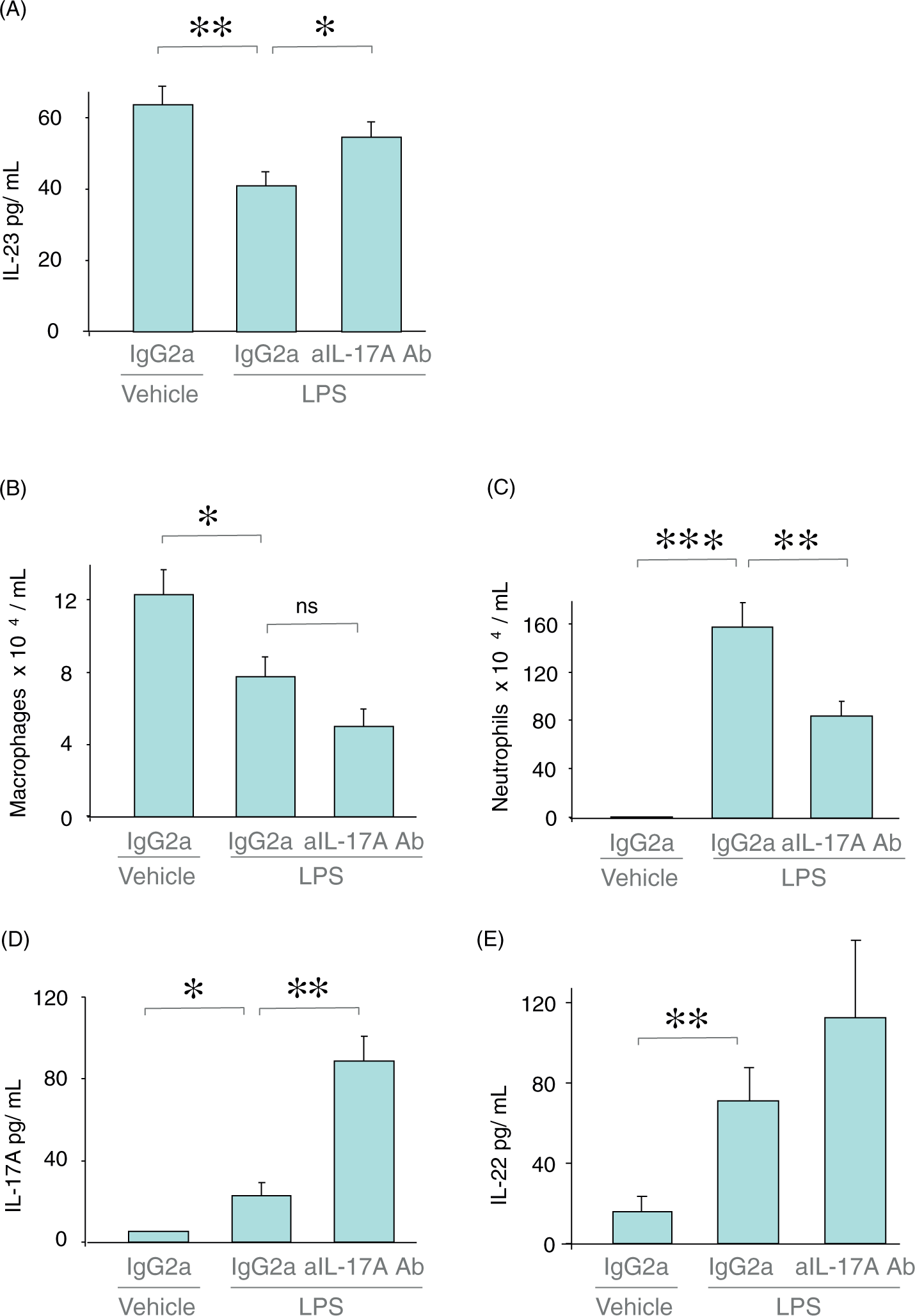
Local IL-23 protein in mouse airways in vivo with and without local stimulation with IL-17A protein
We also performed direct short-term local stimulation with rmIL-17A protein by i.n. instillation without any exposure to LPS. The short incubation time was chosen for the experiments on the effect of rmIL-17A protein to avoid confounding effects of newly recruited neutrophils and monocytes. The BALB/c mice stimulated locally with rmIL-17A protein displayed a significantly decreased concentration of IL-23 protein in cell-free BAL fluid in spite of the mere 2 h of stimulation when compared with the control mice treated with vehicle for IL-17A (Figure 3A). There was no detectable difference in either the concentration of alveolar macrophages (Figure 3B) or neutrophils (Figure 3C) in BAL samples for mice stimulated with rmIL-17A protein compared with the control mice stimulated with vehicle for IL-17A. There was no statistically significant correlation between the concentration of IL-23 and the concentration of BAL macrophages in the BAL samples (Spearman’s rho = −0.176, P = 0.63 for mice treated with IL-17A; Spearman’s rho = −0.43, P = 0.21control mice treated with vehicle for IL-17A) (Supplementary Figure 3).
Extracellular concentrations of IL-23 and alveolar macrophages and neutrophils in BAL samples after local stimulation with recombinant mouse IL-17A protein (rmIL-17A). The BALB/c mice were instilled with 3 µg rmIL-17 protein or its vehicle intranasally. Samples were harvested 2 h later, BAL was performed using 4 × 1.0 ml HBSS (n = 10). n represents the number of mice in each group. (A) Concentration of IL-23 protein in cell-free BAL fluid. (B) Concentration of alveolar macrophages in BAL samples. (C) Concentration of neutrophils in BAL samples. Results are shown as mean with SEM bars. *P < 0.05, ns: P ≥ 0.05.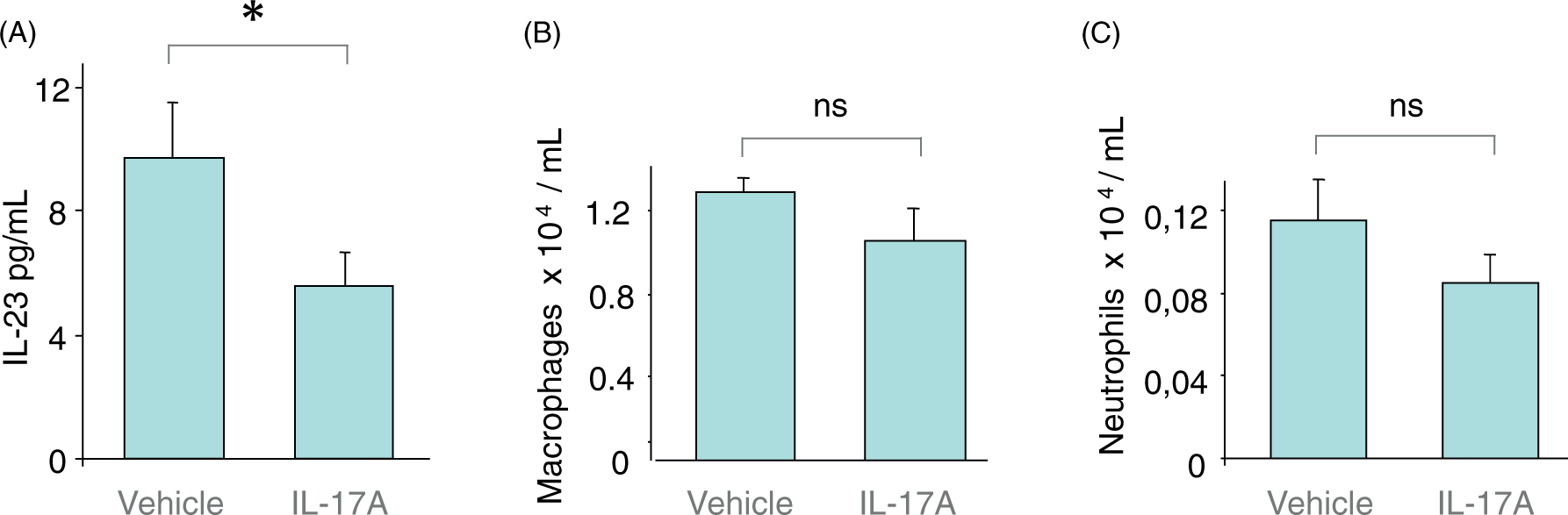
IL-17RA and IL-17RC on human alveolar macrophages
As i.n. instillation of rmIL-17A protein exerted a rapid and reducing effect on the concentrations of IL-23 protein in cell-free BAL fluid, we reasoned that resident alveolar macrophages may play a role. Using freshly isolated human alveolar macrophages and flow cytometry we examined the cell surface expression levels of IL-17RA and IL-17RC (Figure 4A–C). We were able to demonstrate the presence of both receptor units on a series of BAL samples collected from eight human volunteers (Figure 4D).
Expression of IL-17 RA and IL-17RC on human alveolar macrophages harvested from healthy volunteers using BAL, as determined by flow cytometry. (A–C) Representative data from one donor. (A) Representative light scatter plot showing alveolar macrophages in region 1 (R1). (B) R1-gated macrophages stained with equal concentrations of mouse AF647 conjugated anti-human IL-17RA (black) or isotype (grey) and (C) R1 gated alveolar macrophages stained with mouse APC-conjugated anti-human IL-17RC (black) or isotype (grey). Shown in the upper right corner of histograms are the geometric fluorescence values. (D) Cumulative IL-17RA (n = 7) and IL-17RC (n = 8) expression expressed as the geometric mean fluorescence on BAL macrophages. Geometric mean fluorescence values (bar = mean and SD) on the y-axis represent the difference in fluorescence between isotype and specific Ab. n represents the number of BAL donors. FSC, forward scatter; SSC, side scatter.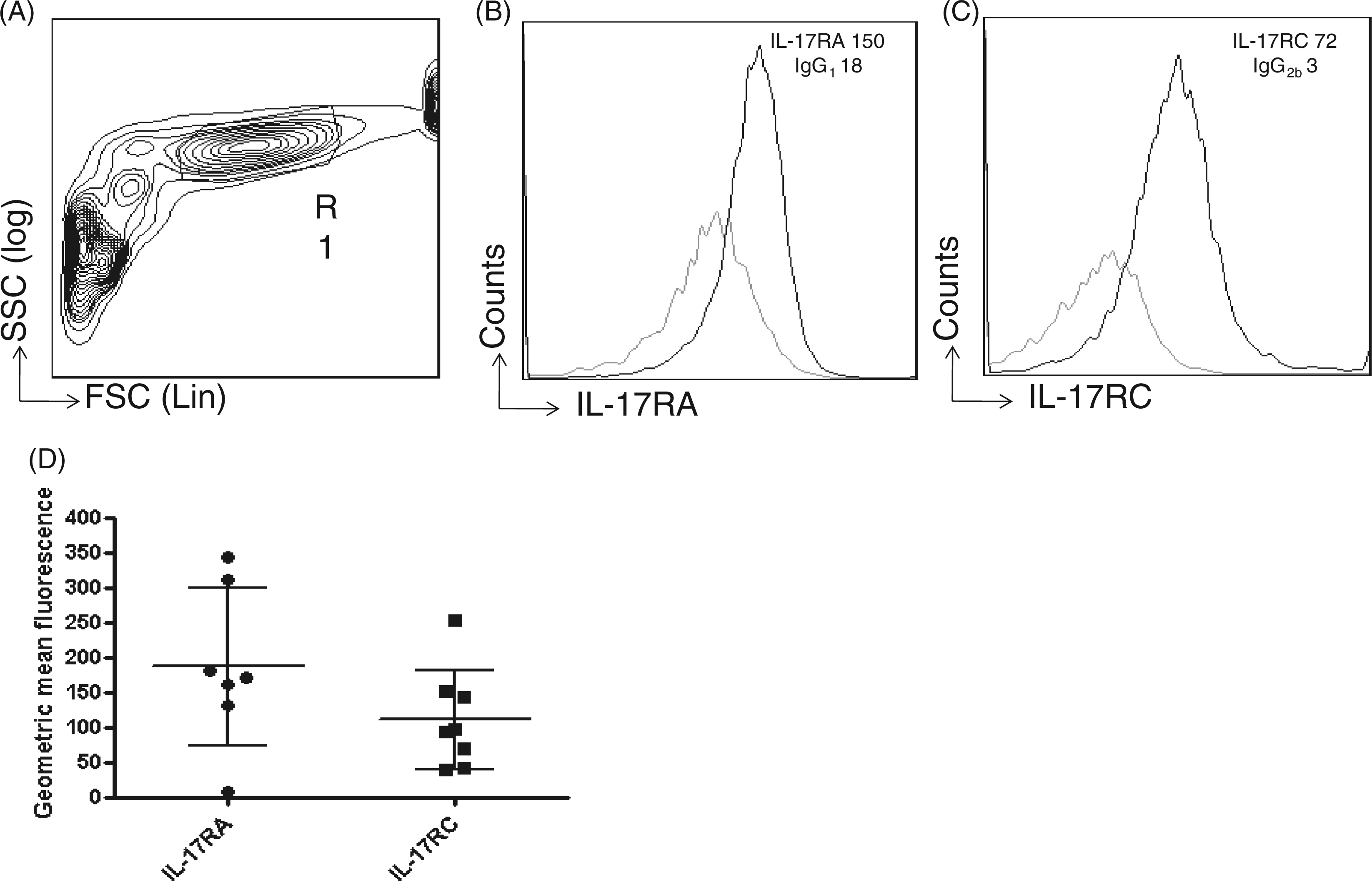
IL-23 from human monocytes and macrophages with and without stimulation by IL-17A protein in vitro
We cultured and stimulated three types of cells from the human monocyte lineage in vitro, namely monocyte-derived macrophages, monocytes and alveolar macrophages. These cells were first activated by exposure to a moderate concentration of LPS (100 ng/ml) and then stimulated by 1 ng/ml of rhIL-17A protein or its vehicle, after which the concentration of IL-23 protein in the conditioned cell culture medium was measured. We found that for all cells not exposed to LPS, IL-23 was not detectable in conditioned medium from the cultures in vitro (data not shown). However, in conditioned medium from human monocyte-derived macrophages exposed to LPS, there was detectable IL-23. In these cells, stimulation with rhIL-17A protein decreased the concentration of IL-23 protein in the conditioned medium; the corresponding IL-23 concentration was consistently higher in the controls treated with the vehicle for IL-17A (Figure 5A). This rhIL-17A-induced decrease in IL-23 protein concentration represented a mean (SEM) relative decrease of 32 (11)% compared with controls. In contrast, in conditioned medium from human monocytes activated by LPS, rhIL-17A protein did not display any corresponding effect on the IL-23 protein concentration (data not shown). These human monocytes also released less IL-23 protein [a mean (SEM) of 85 (33) pg/ml, n = 4] after exposure to LPS compared with the monocyte-derived macrophages [a mean of 185 (43) pg/ml, n = 10]. Moreover, we also tested whether rhIL-17A could influence the IL-12 protein concentration caused by LPS exposure in the human monocyte-derived macrophages cultured in vitro. However, we found no detectable concentrations of IL-12 protein in the conditioned medium from these cells (Supplementary Figure 4).
Extracellular concentrations of IL-23 protein and IL-23 mRNA in human monocyte-derived macrophages and alveolar macrophages harvested from blood or BAL samples and cultured in vitro. n represents the number of BAL or blood donors. Each donor contributed to one independent experiment. Concentration of IL-23 protein in conditioned medium from (A) monocyte-derived macrophages (n = 10) and (B) alveolar macrophages (n = 6), all exposed to LPS (100 ng/ml) and stimulated either with rhIL-17A (1 ng/ml) or its vehicle (supplemented medium alone) over 24 h. Results are shown as individual values. Messenger RNA for the IL-23 p19 or p40 subunits measured from (C) monocyte-derived macrophages (n = 11), and (D) alveolar macrophages (n = 5) activated by LPS (100 ng/ml) and rhIL-17A (1 ng/ml) or its vehicle (supplemented medium alone) over 24 h. Results are shown as mean with SEM bars. *P < 0.05.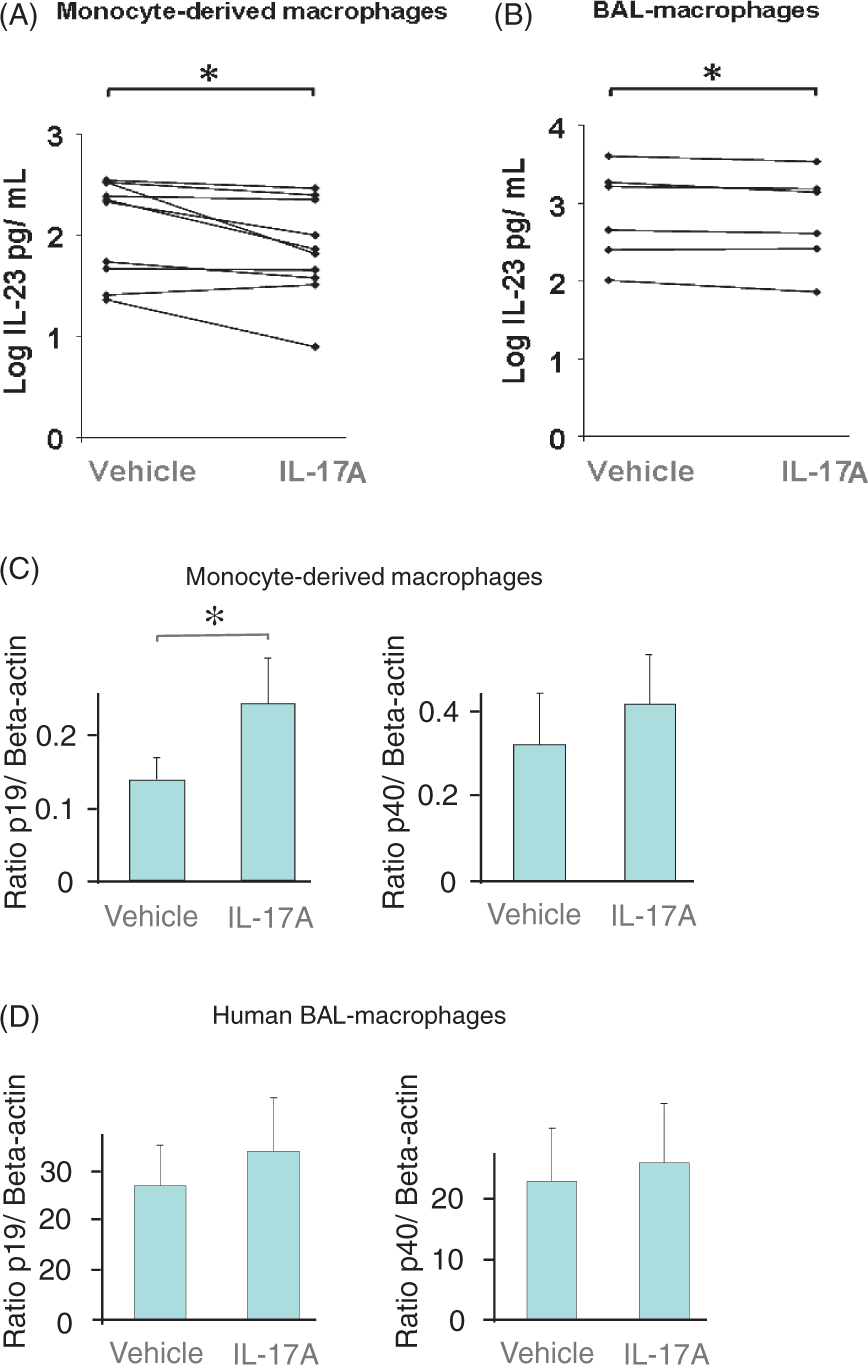
Interestingly, very similar to what was observed for the monocyte-derived macrophages, rhIL-17A stimulation decreased the concentration of IL-23 protein in the conditioned medium from human alveolar macrophages exposed to LPS (Figure 5B). This rhIL-17A-induced decrease in IL-23 protein concentration represented a mean (SEM) relative decrease of 15 (5)% compared with the matching controls.
Messenger RNA for IL-23 in human macrophages with and without stimulation by IL-17A protein in vitro
Stimulation with rhIL-17A protein further increased mRNA for the IL-23 subunit p19 in the human monocyte-derived macrophages exposed to LPS (Figure 5C). Although the same trend was found for the mRNA for the IL-23 subunit p40 in the monocyte-derived macrophages exposed to LPS (Figure 5C), it was not statistically significant. In analogy, stimulation with rhIL-17A protein tended to increase both p19 and p40 mRNA in human alveolar macrophages exposed to LPS (Figure 5D), even though these effects were not proven statistically significant.
The inhibition of Rac1 and concentrations of IL-23 protein from human macrophages with stimulation by IL-17A protein in vitro
In contrast to the hypothesized outcome, the specific inhibitor of the GTPase Rac1, NSC23766 caused a decrease of the IL-23 concentration in the conditioned medium from the IL-17A-stimulated monocyte-derived macrophages from humans that were exposed to LPS (Figure 6). This unexpected effect of the Rac1 inhibitor was statistically significant (Figure 6). There was also a similar trend in cells not stimulated by rhIL-17A protein, even though this trend did not reach statistical significance (data not shown).
Extracellular concentrations of IL-23 protein in conditioned medium from human monocyte-derived macrophages, all exposed to LPS (100 ng/ml) and stimulated with hIL-17A (1 ng/ml) with and without pre-treatment using the Rac1 inhibitor (NSC23766: 50-300 µM) or its vehicle (supplemented medium alone) over 24 h (n = 5). n represents the number of blood donors. Each donor contributed to one independent experiment. Results are shown as mean with SEM bars. *P < 0.05.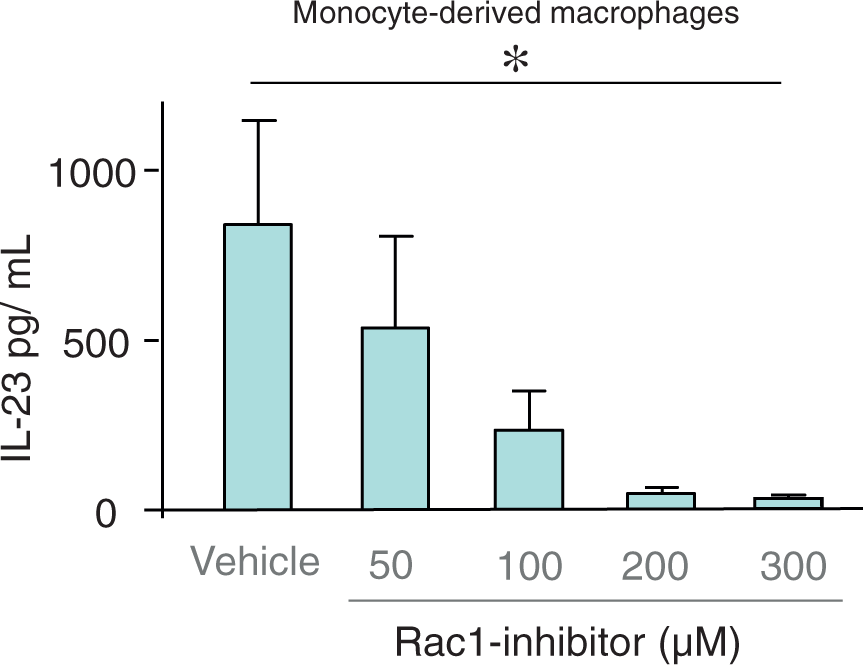
Discussion
This study employed three different experimental in vivo models, including two different mouse strains, comprising i.v. inoculation of the Gram-positive bacteria, S. aureus and i.n. administration of LPS, a component from the Gram-negative bacteria E. coli, as well as recombinant protein for the cytokine of interest per se. The results from these in vivo models indicate that IL-17A decreases the extracellular concentration of its critical upstream regulator IL-23 during pulmonary inflammation. Confirmatory results were obtained from three experimental models in vitro—from isolated monocytes, human monocyte-derived macrophages and alveolar macrophages.
Hypothetically, there are two confounding factors that could influence the IL-17A-induced decrease in extracellular IL-23 protein in vivo. The first hypothetical factor is the possibility that IL-17A consistently decreases the number of IL-23-producing macrophages in the lungs. However, when carefully examining the results of our in vivo experiments, we found no consistent IL-17A-associated alteration of the concentrations of BAL macrophages that related to levels of IL-23 protein in the three different models that we utilized. Moreover, when examining the results of our in vitro experiments, where a fixed number of human monocyte-derived or alveolar macrophages were exposed to LPS, we did find that stimulation with rhIL-17A protein substantially decreases the concentration of IL-23 protein in the cell-free, conditioned medium, thus confirming the inhibitory action of IL-17A protein on the extracellular concentration of IL-23 protein that was indicated in vivo. The second hypothetical factor is the possibility that IL-17A protein decreases the viability of the macrophages. However, our group has previously shown that stimulation with recombinant mouse IL-17A protein (1 ng/ml, which is the same concentration as used in the current in vitro experiment) prolongs the survival of mice BAL macrophages cultured for 20 h in vitro in a statistically significant manner. 47 Altogether, this provides evidence for a direct effect on macrophages. Thus, the decrease of extracellular IL-23 protein in the lungs cannot be attributed merely to an IL-17A-induced decrease in the number or viability of macrophages.
Notably, we here demonstrate for the first time that human alveolar macrophages from healthy, non-smoking volunteers do express IL-17 receptor protein for both IL-17RA and IL-17RC. This is an important functional prerequisite for the results from our experiments on the release of IL-23 protein in the airways in vivo and on macrophages in vitro. It is true that the receptor components IL-17RA and IL-17RC have previously been demonstrated at the protein level in monocytes from human blood48,49 and at the mRNA level in macrophages from mouse lungs, but never before in human alveolar macrophages. 50 Moreover, in our current experiments, IL-17A did not alter the extracellular release of IL-12 protein from the human monocytes and monocyte-derived macrophages in vitro. Indeed, this finding is in line with previous studies, demonstrating that alveolar macrophages stimulated with LPS do not release IL-12.51,52 It thus seems as if the macrophages require stimulation with both LPS and IFN-γ in order to release IL-12 protein. The fact that there is no IL-12 protein in the conditioned medium for cell cultures argues that the IL-23 signal measured by ELISA in our study is not due to cross-reactivity with IL-12 protein.
The fact that stimulation with rhIL-17A did not decrease IL-23 p19 mRNA in monocyte-derived macrophages or in alveolar macrophages from humans in vitro, and that exogenous rmIL-17A decreased the concentration of IL-23 protein in cell-free BAL-fluid from mice as early as 2 h later, suggests one of two sites of action: the inhibitory action of IL-17A on extracellular IL-23 could be located either at the post-transcriptional or at the post-translational level, or at both levels. This type of discrepancy between p19 mRNA and extracellular IL-23 protein, as well as the fact that p19 mRNA does not necessarily always match the presence of IL-23 protein, has been demonstrated previously in at least two studies.53,54 In previous publications, it has also been suggested that IL-23 signaling can be inhibited both at the post-transcriptional and post-translational level, in line with our current findings. An alternatively spliced soluble variant of the IL23Rα-chain blocks soluble IL-23,55,56 and the anti-inflammatory drug celecoxib inhibits IL-23 by increasing the delivery of IL-23 to the proteasome. 57
We briefly addressed the potential coupling of the Rho family GTPase Rac1 58 to intracellular signaling downstream of IL-17 RA and IL-17RC, and to the regulation of IL-23 protein in our experiments.37,38 The rationale for this was twofold: first, stimulation with IL-17A protein is known to increase Rac1-activation in human mesenchymal stem cells;37 second, the inhibition of Rac1, using over-expression of dominant-negative Rac1 (N17Rac1), does increase mRNA for IL-23 p19 in human monocyte-like THP-1 cells exposed to LPS. 38 In our study we used NSC23766, a specific inhibitor of Rac1, in human monocyte-derived macrophages exposed to LPS to evaluate the involvement Rac1 in mediating the inhibitory effect of IL-17A on the extracellular concentration of IL-23 protein. However, we obtained no evidence that Rac1 mediates the specific inhibitory effect of IL-17A protein on extracellular IL-23 protein. On the contrary, the Rac1-inhibitor tended to decrease the extracellular concentration of IL-23 protein. Notably, the Rac1-inhibitor did this in a concentration-dependent manner with and without stimulation using hrIL-17A, even though this decreasing effect reached statistical significance only in cells stimulated by IL-17A. We now forward two feasible explanations for what might appear like a discrepancy between our results and those of the preceding study; the study suggesting that the inhibition of Rac1 increases mRNA for IL-23 p19 in human monocyte-like human acute monocytic leukemia cell line (THP-1) cells exposed to LPS. 38 One explanation is that they only found increased IL-23 p19, but not IL-23 p40, so that the net IL-23 protein does not necessarily need to be increased. Another explanation is that in the preceding study, only p19 mRNA was measured; p19 or IL-23 protein was never assessed. As stated earlier in this discussion, p19 mRNA does not always match the presence of IL-23 protein, so although p19 mRNA was increased, the corresponding p19 and IL-23 protein may not have been increased in the referred study.53,54
Because the highest utilized concentration of the Rac1-inhibitor abrogated the measurable levels of IL-23 almost completely in our experiments, the most straight-forward conclusion is that Rac1 is involved in the release of IL-23 protein in human macrophages, but not specifically in the inhibitory effect exerted by IL-17A protein on macrophages exposed to LPS. Indeed, this conclusion is supported by several previous studies.59–63 In these previous studies, Rac1 was found to mediate the release of cytokines such as IL-5, IL-6, IL-7, IL-8, IL-13, G-CSF, GM-CSF, MIP-1α, IFN-γ and IL-1β from human endothelial cells, peripheral blood monocytes and THP-1 cells, a human monocytic cell line.5, 6, 10,59–63 Interestingly, Rac1 seems to achieve its releasing effect on cytokines through its effects on the actin cytoskeleton.12,64,65 The form of exocytosis that is dependent of Rac1 is termed ‘regulated exocytosis’ and involves secretory granules or vesicles containing preformed cytokines that can be mobilized to the cell surface for release.62–66 It therefore seems feasible that the Rac1-specific inhibitor NSC23766 used in our study actually decreased the release of IL-23 by reducing exocytosis.
Here, we also characterized the downstream immunological consequences of the IL-17A-induced inhibition of the extracellular release of IL-23 in vivo by assessing the local protein concentration of mouse IL-17A and IL-22, respectively—two cytokines normally being produced in response to the activation of the IL-23-receptor.24,29,67 Interestingly, we found an increased concentration of IL-17A protein in the group of mice that were exposed to LPS and pre-treated with the aIL-17A Abs compared with the corresponding mice pre-treated with the isotype control Abs. The result of the performed interference test (see Materials and methods) assured us that the presence of the aIL-17A Ab or its isotype control IgG2a was not markedly affecting the concentration of mouse IL-17A protein in the BAL samples detected by the utilized ELISA kit. We therefore conclude that the detected concentration of mouse IL-17A protein in the BAL samples from the LPS-exposed mice pre-treated with aIL-17A Ab was either accurate or, possibly, somewhat underestimated. We find it important to point out that the concentration of neutrophils in the BAL samples was significantly decreased after LPS exposure plus pre-treatment with aIL-17A Ab compared with LPS exposure plus treatment with IgG2a. This finding confirms that endogenous IL-17A was neutralized and non-functional owing to the treatment with aIL-17A Ab. That recruitment of neutrophils is dependent on IL-17 and is reduced when IL-17 is non-functional has been demonstrated in numerous previous studies before.6,8,68 Thus, mice exposed to LPS and pre-treated with isotype control Ab that have functional IL-17A have decreased extracellular concentrations of IL-23 protein, while the mice exposed to LPS and pre-treated with anti-IL-17A Ab that neutralizes functional IL-17A, have substantially higher extracellular concentrations of IL-23 protein. Importantly, this is fully in line with the conclusion that functional IL-17A protein and signaling is required for the down-regulation of the concentration of the upstream regulator IL-23 at a certain, critical time point during inflammation. In further support of this conclusion: two previous studies have shown an increase in mRNA expression and protein secretion for mouse IL-17A, as well as in the number of IL-17A-producing neutrophil-regulatory T cells (CD4−CD8−αβlow) in mice deficient in IL-17RA.68,69 When we addressed the functional consequences for the immunological signaling downstream of the IL-23 receptor in vivo in mice exposed to LPS i.n., we detected a trend towards increased extracellular concentration of IL-22 protein in the group of mice that had been exposed to LPS and pre-treated with aIL-17A Ab compared with the mice that were exposed to LPS and pre-treated with an isotype control Ab. Unfortunately, this trend did not reach statistical significance with the limited size of our material. However, also in line with our current results, it has recently been suggested that there is a pathway for IL-17A-mediated inhibition of Th17 cell effector cytokine expression, based upon the finding that the absence of endogenous IL-17A protein leads to an increase in IL-22 protein in yet another mouse model of pulmonary inflammation 70 and an increase in IL-22 mRNA in a mouse model of colitis. 13 It has also recently been shown in mouse cells in vitro that IL-17A protein suppresses the secretion of IL-22 protein from Th17 cells, 70 as well as decreasing IL-22 mRNA in splenocytes 69 and Th17 cells. 71
In conclusion, we here forward functional in vivo and in vitro evidence that, during pulmonary inflammation, endogenous, as well as exogenous, IL-17A protein exerts an inhibitory effect on the extracellular release of IL-23 protein, the upstream regulator of IL-17A-producing lymphocyte subsets. Presumably, this negative feedback is not located at the transcriptional level. It seems more likely that it is located at the post-transcriptional and/or post-translational level in alveolar macrophages. We thus propose that alveolar macrophages constitute critical cells for the demonstrated feedback mechanism, enabled by their co-expression of IL-17RA and IL-17RC—receptors here demonstrated in primary human alveolar macrophages for the first time. We suggest that Rac1 is involved in the release of IL-23 from human macrophages, but not specifically in mediating the inhibitory effect of IL-17A per se. Based upon the current findings, it can be speculated that a negative feedback mechanism in alveolar macrophages protects from excessive activity in IL-17A-producing lymphocytes, including Th17 cells, during pulmonary inflammation. The main purpose with this mechanism may be to limit the potentially tissue-damaging impact of antibacterial host defense once it is triggered. The demonstration of this feedback mechanism warrants future investigations of its relevance for human lung disorders with increased IL-17A signaling, such as asthma and cystic fibrosis.72–75
Footnotes
Funding
Research funding was obtained from the Swedish Heart-Lung Foundation (project #20100290), Swedish Research Council (project #K2008-57X-0948-19-3), King Gustav V’s & Queen Victoria’s Freemason Foundation and Federal Funding at Karolinska Institute.
Acknowledgements
The authors wish to thank the clinical team at the Royal Liverpool and Broadgreen University Hospitals NHS trust for their technical expertise and support. The authors also acknowledge the Liverpool NIHR Biomedical Research Centre high throughput screening facility for infrastructural support in this project.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.