
Abstract
Mycobacterium avium ssp. paratuberculosis (MAP) causes Johne’s disease, a chronic, granulomatous enteritis of ruminants. Dendritic cells (DC) of the gut are ideally placed to combat invading mycobacteria; however, little is known about their interaction with MAP. Here, we investigated the interaction of MAP and the closely related M. avium ssp. avium (MAA) with murine DC and the effect of infected macrophages on DC maturation. The infection of DC with MAP or MAA induced DC maturation, which differed to that of LPS as maturation was accompanied by higher production of IL-10 and lower production of IL-12. Treatment of maturing DC with supernatants from mycobacteria-infected macrophages resulted in impaired DC maturation, leading to a semi-mature, tolerogenic DC phenotype expressing low levels of MHCII, CD86 and TNF-α after LPS stimulation. Though the cells were not completely differentiated they responded with an increased IL-10 and a decreased IL-12 production. Using recombinant cytokines we provide evidence that the semi-mature DC phenotype results from a combination of secreted cytokines and released antigenic mycobacterial components of the infected macrophage. Our results indicate that MAP and MAA are able to subvert DC function directly by infecting and indirectly via the milieu created by infected macrophages.
Introduction
Macrophages and dendritic cells (DC) are important elements of both innate and adaptive immune responses. As antigen presenting cells (APCs) they play a central role in processing and presenting microbial Ags to T cells. During mycobacterial infection APCs are essential for inducing a protective immune response. 1 In addition to their role as APCs, macrophages are the primary target cells for mycobacterial persistence during mycobacterial infection. Thus, pathogenic mycobacterial species have developed mechanisms that subvert macrophage effector functions, which, in turn, favours intracellular survival.2–4
DC are specialized in processing and presenting Ags for which they have to differentiate to a highly competent APC. This maturation process is influenced by the micro-environment created by other immune cells and microbial pathogens. Thus, immature DC are present in peripheral tissues and are very effective in taking-up Ags, but are still only weakly able to initiate a primary T cell response. 5 After exposure to inflammatory stimuli, such as cytokines and microbial products, differentiation into mature DC occurs. Mature DC are highly professional APCs, able to take up Ags and migrate into the lymphoid organs where they activate naïve T cells. 6 Mature DC are characterized by an enhanced expression of type I and II molecules of the MHC and co-stimulatory molecules (CD80, CD86, CD40), which enable them to prime naïve T cells. 7 In addition to their role as APCs, DC can direct the T cell response towards Th1 or Th2 phenotypes. Thus, the capacity of DC to produce IL-12 and IFN-α that can enhance the secretion of IFN-γ by T cells and NK cells contributes to perpetuation of a Th1-biased response.8–10
Likewise, a Th1-dominated immunity, mediated by IFN-γ-secretion of CD4+ T cells, represents the major protective immune response against pathogenic mycobacteria, such as Mycobacterium tuberculosis and Mycobacterium bovis.11,12 The critical role of DC in this process was emphasized by the work of Tascon et al. 5 who established that transfer of M. tuberculosis-infected DC confers protective anti-mycobacterial immunity in a murine model of experimental M. tuberculosis infection. Furthermore, the depletion of CD11c+ cells (including DC) in mice before an i.v. infection with M. tuberculosis delays the development of CD4+ T cell responses and results in an impaired immune control of M. tuberculosis. 13 To obtain a Th1-biased immunity after an infection with mycobacteria, DC need to mature properly and up-regulate their IL-12 production profoundly. 8
The three genetically most closely related Mycobacterium avium ssp., M. avium ssp. avium (MAA), M. avium ssp. hominissuis and M. avium ssp. paratuberculosis (MAP), are found ubiquitously in the environment and may cause mycobacteriosis in animals, as well as in immunocompromised humans.14–17 In addition, MAP is pathogenic for ruminants, causing a chronic, non-treatable granulomatous enteritis known as Johne’s disease (paratuberculosis), 18 which occurs worldwide with increasing incidence. Additionally, MAP is discussed as being the etiological agent of Crohn’s disease in humans.19,20
All M. avium ssp. use the intestine as their preferred port of entry. After crossing the mucosal barrier via enterocytes or M-cells, mycobacteria will be recognized by subepithelial macrophages, as well as intestinal DC. While macrophages are clearly defined as target cells for mycobacterial persistence, the role of DC in mycobacterial infection is still unclear.
M. avium ssp. differ in their capacity to activate macrophages. We found previously that MAP is able to survive in macrophages and restrict the macrophage pro-inflammatory response more efficiently than MAA.3,21 Furthermore, only MAP-infected macrophages were able to inhibit the Ag-specific stimulation of a CD4+ T-cell line. 22
In the present study we analysed the capacity of MAP and MAA to induce maturation of murine bone marrow-derived DC and to modulate their cytokine expression. Moreover, we investigated the capacity of MAP- or MAA-infected macrophages to interfere with DC maturation and activation.
Materials and methods
Reagents
If not stated otherwise, all chemicals and LPS, purified from Escherichia coli 0127:B8, were purchased from Sigma (Munich, Germany). Mycobactin J was purchased from Synbiotics (Stuttgart, Germany), recombinant-mouse granulocyte macrophage-colony stimulating factor (rmGM-CSF) from R&D Systems (Minneapolis, MN, USA). All Abs were obtained from BD Biosciences Pharmingen (Hamburg, Germany). Recombinant cytokines (IL-1β, IL-6 and IL-10) were purchased from Miltenyi (Bergisch Gladbach, Germany).
Bacterial strains and culture
The MAP strain DSM 44135 and the MAA strain DSM 44158 (ATCC19421 = NCTC8559) were cultured, heat-inactivated and prepared for infection as described previously. 3 These strains were selected for this study as they were shown previously to differentially affect macrophage effector functions.3,21,22
Macrophage cell culture
The murine macrophage-like cell line J774.A1 (ATCC, TIB-67) was maintained and infected, as described by Kuehnel et al. 3 In brief, 3 d before infection 2 × 106 macrophages were seeded in antibiotic-free cell culture medium on 9-cm diameter cell culture dishes. For infection, medium was removed and the cells were incubated with a suspension containing mainly single mycobacteria (MAA, MAP) of an OD of 0.15 at 660 nm (OD660) in antibiotic-free medium for 2 h, which resulted in a multiplicity of infection (MOI) of 10:1. Then, the medium was removed, cells were washed with PBS to remove non-ingested bacteria and fed with 10 ml fresh antibiotic-free, serum-free medium. After 24 h supernatants (SN) were harvested and filtered through 0.22 -µm filters. In addition, SN from non-infected macrophages were obtained and treated similarly.
In vitro generation of bone marrow-derived murine DC
DC were generated from mouse bone marrow, according to Lutz et al. 23 In brief, femurs were removed from BALB/c mice (Charles River) aged 8–20 wk. Both ends were cut off and the bone marrow flushed with PBS. Cells were singularized by vigorous pipetting. Thereafter, the suspension was centrifuged at 300g and the pelleted cells were suspended in 5 ml RPMI medium 1640 (Biochrom, Berlin, Germany) supplemented with 1% penicillin/streptomycin (PAA, Pasching, Austria), 10% FCS (PAA) and 50 µM 2-mercaptoethanol (complete medium). Cells were seeded at a density of 2.5 × 105 per well in a 12-well plate (Greiner, Frickenhausen, Germany) in 1 ml complete medium containing rmGM-CSF (20 ng/ml). Then, cells were cultured at 37℃ and 5% CO2. After 3 d, 1 ml of complete medium containing rmGM-CSF (20 ng/ml) was added and on d 6 and d 8 half of the culture SN was removed, centrifuged, the pellet suspended in complete medium containing rmGM-CSF (20 ng/ml) and returned to the cell suspension. After 9 d of differentiation, DC were transferred into a 12-well plate in complete medium. The cells were treated with live or heat-killed MAP or MAA at a MOI of 10 for 24 h. DC maturation and cytokine production was compared with cells stimulated with 0.375 µg/ml LPS for 24 h. Viability was tested by annexin V-FITC and propidiumiodid (PI) from BD Biosciences Pharmingen according to the manufacturers instructions. The amount of viable cells was 90–95%; after LPS stimulation it was >65%.
Maturation of DC in the presence of SN from mycobacteria-infected macrophages
To determine the effect of mycobacteria-infected macrophages on DC maturation, cells were cultured as described above in the presence of SN from MAP- or MAA-infected macrophages, or from non-infected macrophages designated as MΦ-MAP-SN, MΦ-MAA-SN, and MΦ-control-SN respectively. For this, after 3 d of culture in the presence of rmGM-CSF-supplemented complete medium, cells were cultured in complete medium containing rmGM-CSF only or with medium containing rmGM-CSF plus 10% (v/v) of cell-free SN. Ten% v/v SN was the lowest volume with highest suppressive activity and lowest cytotoxicity as determined by dose response (data not shown). On d 6 and 8, cells were fed with fresh complete medium containing rmGM-CSF and the macrophage culture SN. On d 9 cells were stimulated with LPS (0.375 µg/ml) for 24 h to induce final maturation. The concentration of 0.375 µg/ml was chosen as it induced robust final maturation of DC with lowest cytotoxic effects (data not shown). As a positive control, non-treated control cells were stimulated at d 9 with LPS for 24 h.
Flow cytometry
Twenty-four h post-infection or stimulation cells were harvested and cytometric analysis was performed by using a FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA) as described previously. 24 To evaluate phenotypic DC maturation, the following Abs were used: biotin anti-mouse I-A/I-E, FITC anti-mouse CD86, phycoerythrin anti-mouse CD11c, streptavidin-peridin chlorophyll-a protein (SAv-Per CP) conjugate. Analysis was performed on Coulter Epics XL using Expo 32 ADC software (Beckman Coulter, Miami, FL, USA).
Mixed leukocyte reaction
T-Lymphocytes were isolated from spleen of NMRI (Naval Medical Research Institute) mice (Charles River, Sulzfeld, Germany) and used for mixed leukocyte reaction, as described previously. 25 In brief, T cells isolated from spleen were stained with carboxyfluorescein diacetate succinimidylester (CellTrace, Invitrogen, Eugene, OR, USA) for 10 min at 37℃, washed and suspended in complete medium. DC collected after 24 h of infection or stimulation were mixed with T cells in a 96-well round bottom plate (Greiner, Frickenhausen, Germany) at a ratio of 1:20. The mixed cells were incubated at 37℃ and 5% CO2 for 5 d.
ELISA analysis
Cell culture SN of J774.A1 macrophages or DC were collected 24 h post-infection. IL-1 β, IL-6, IL-10, IL-12 and TNF-α were assayed by ELISA, as described by the manufacturer (R&D Systems, Minneapolis, MN, USA).
Western blot analysis
SDS-PAGE was performed with a 4% stacking and a 12% separating gel. SN of untreated or infected J774.A1 cells were precipitated with trichloroacetic acid (10%) overnight, diluted in a simple loading buffer and heated to 100℃ for 5 min. Gels were either stained with Coomassie brilliant blue or used for immunoblotting onto polyvinylidene fluoride membranes. After blocking with 5% skimmed milk, membranes were incubated with bovine serum of MAP-infected cows, washed and developed with peroxidase conjugated anti-bovine immunoglobulins and chemiluminescence.
Statistical analysis
The results are presented as mean ± SEM. Statistical significance was determined by a one-way ANOVA followed by post hoc test (Dunnett’s) or two-tailed paired t-test when only two groups were compared with each other.
Results
Infection with MAP and MAA induces maturation of DC
In the first set of experiments we analysed the interaction of MAA and MAP with DC. According to Lutz et al., 23 differentiation of DC from the bone marrow by rmGM-CSF is completed after a final maturation evoked by a bacterial stimulus, such as LPS and is indicated by enhanced levels of CD86 and MHCII, as well as an enhanced capacity to activate T cells.
To determine whether infection with MAP and MAA leads to maturation of DC cells were infected with live MAP or MAA at MOI 1:1 (MOI 1), 5:1 (MOI 5) or 10:1 (MOI 10). Additionally, cells were treated with heat-killed MAP or MAA. Successful bacterial uptake was confirmed by electron microscopic evaluation (data not shown). The expression of CD86 and MHCII on CD11c+ cells was determined 24 h post-exposure and compared with Ag expression of untreated DC as a negative control (nil) and LPS-stimulated DC as positive control. As shown in Figure 1A, DC expression of CD86 and MHCII was increased after infection with MAP and MAA when compared with untreated cells. Expression levels similar to LPS-stimulated DC could be induced with MOI of 5:1 and 10:1 (Figure 1A). There were no significant differences in expression of these markers between infection with live bacteria and treatment with heat-killed bacteria (Figure 1A, B).
DC maturation after infection with MAP and MAA. Bone marrow-derived DC were infected with live or treated with heat-killed (dead; striped bars) MAP and MAA at indicated MOI or were stimulated with LPS at d 9 or left untreated (nil). Expression of CD86 (A) and MHCII (B) was measured 24 h post infection. Means ± SEM from four experiments are shown. (C) Mixed leukocyte reaction of infected DC and T cells. For examination of T cell proliferation untreated (nil), LPS-stimulated (LPS) or mycobacteria-infected (MAP, MAA) DC were incubated for another 5 d with carboxyfluorescein diacetate succinimidylester-stained T lymphocytes. Percentage of proliferated T cells is shown as means ± SEM from three experiments (*P < 0.05).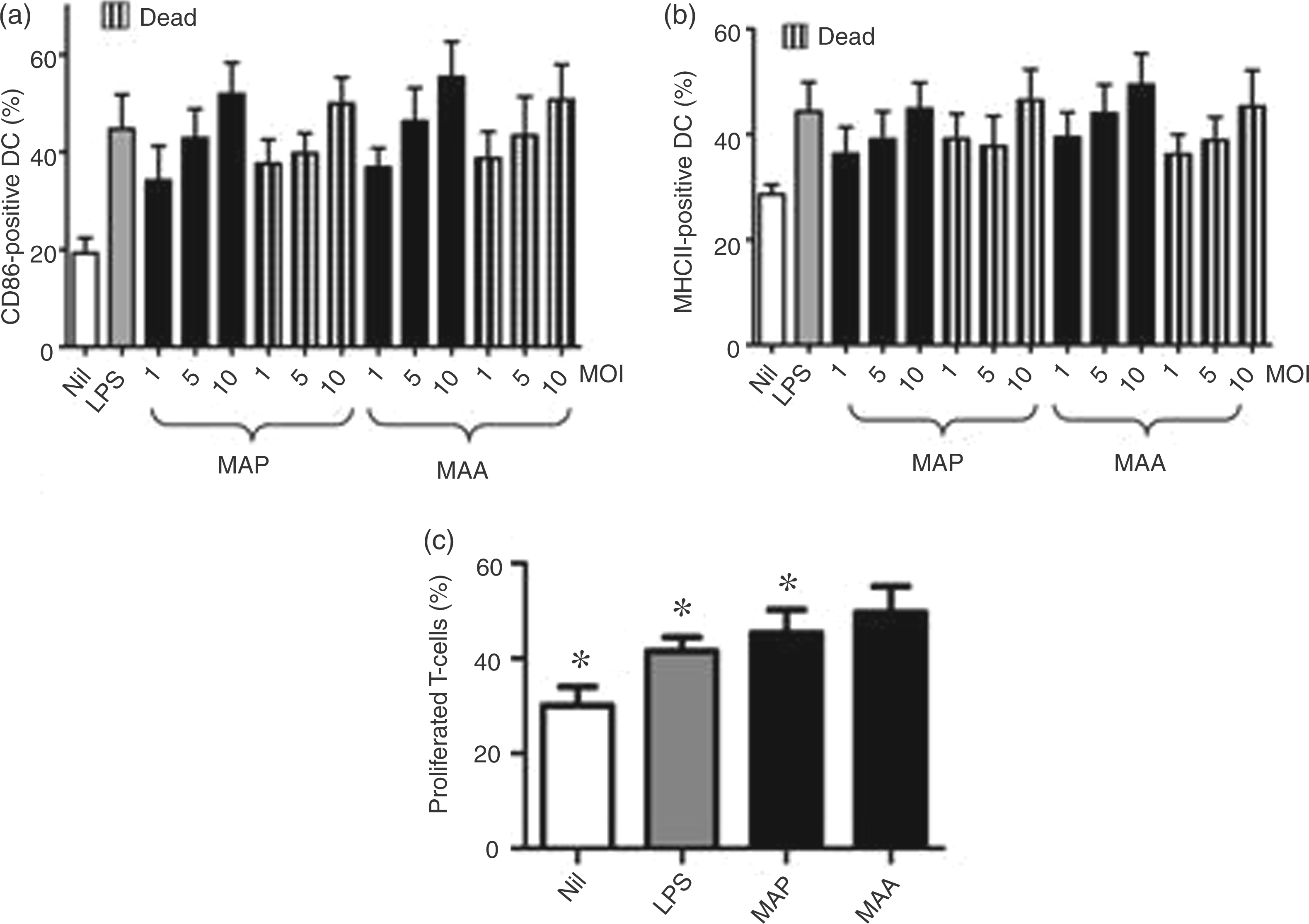
The ability of MAP- and MAA-infected DC to induce T cell activation was assessed in a mixed leukocyte reaction (MLR). Twenty-four h after treatment of DC with live or heat-killed bacteria (MOI 10:1) cells were incubated with murine spleen T lymphocytes for 5 more d and proliferation of T cells was determined by FACS. Similar to LPS-treated cells, both viable (Figure 1C) and heat-killed (data not shown) MAP- or MAA-infected DC were able to increase T cell proliferation in comparison with non-infected control cells. The percentage of proliferated T cells after incubation with MAP- or MAA-infected DC and LPS-maturated DC was approximately twofold higher than after incubation with untreated control DC. In all experiments the T cell proliferation induced by MAA-infected DC slightly exceeded the proliferation induced by MAP-infected DC.
Infection of DC with MAP and MAA induces IL-10 and IL-12 production
Subsequently, we tested the ability of DC maturated by MAP or MAA to produce IL-10 and IL-12. Compared with untreated control cells and DC stimulated with LPS, the infection of DC with MAP and MAA (MOI 10:1) significantly increased IL-10 secretion (Figure 2). Furthermore, MAP and MAA induced IL-12 production, but to a significantly lower extent than LPS-stimulated DC. Infection with live bacteria induced stronger secretion of both IL-10 and IL-12 compared to heat-killed MAP and MAA. In contrast, the expression of the pro-inflammatory cytokine TNF-α was induced to the same extent by viable and heat-killed mycobacteria, but the level of TNF-α was significantly higher than that released by LPS stimulated DC (Figure 2).
IL-10, IL-12 and TNF-α production of MAP- and MAA-infected DC. Bone marrow-derived DC were infected with live or treated with heat-killed (dead) MAP and MAA at MOI 10:1 or stimulated with LPS for 24 h. Cytokines in supernatants were detected by ELISA for IL-10, IL-12 and TNF-α. Means ± SEM from nine experiments are shown (***P < 0.001; **P < 0.01; *P < 0.05).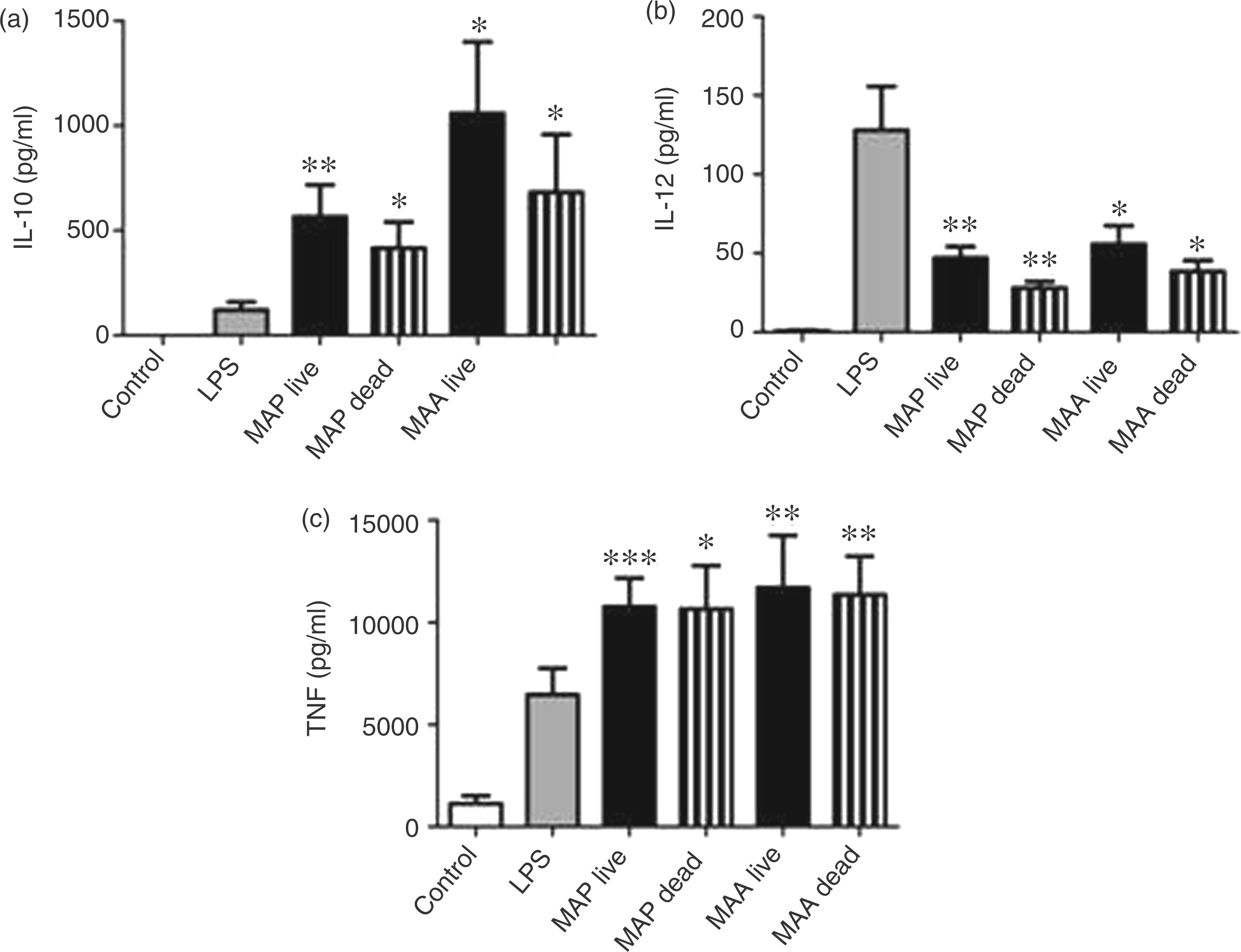
MAP- and MAA-infected macrophages are able to impair DC maturation
Unambiguously, the primary target cell for MAP is the macrophage. Thus, an impaired macrophage response might influence DC maturation. In a second set of experiments we therefore tested the capacity of MAP-infected macrophages to modulate DC maturation using SN from viable MAP-infected macrophages. For these experiments we used J774.A1 macrophages as we have previously shown that MAP and MAA can survive within these macrophages for up to 5 d without affecting cell integrity. 22
The influence of macrophage SN on DC maturation was monitored as described in the Materials and methods. Stimulation of control cells and cells cultured in the presence of control-MΦ-SN with LPS increased expression of CD86 and MHCII on CD11c+ cells. After incubating with MAP-MΦ-SN or MAA-MΦ-SN, a significantly lower expression of CD86 and MHCII expression was observed following LPS treatment, which was similar to that of non-treated DC. Compared with the LPS-treated controls, there were 2.1-fold fewer CD86-positive cells after treatment with MAP-MΦ-SN and 3.9-fold fewer CD86-positive cells after incubation with MAA-MΦ-SN (Figure 3A). The percentage of MHCII-positive DC was 1.8-fold lower after administration of MAP-MΦ-SN and 3.8-fold lower after MAA-MΦ-SN treatment (Figure 3B).
Impaired DC maturation after incubation with SN from MAP- or MAA-infected macrophages. Bone marrow-derived cells were cultured in complete medium with rmGM-CSF. At d 3, 6 and 8 cells were treated with cell-free SN from non-infected macrophages (control-MΦ-SN) or from macrophages infected with viable MAP- or MAA (MAP-MΦ-SN, MAA-MΦ-SN), as described in Materials and methods. Cells cultured in the presence of rmGM-CSF only served as controls for LPS-induced final DC maturation. At d 9 DC were stimulated with 0.375 µg/ml LPS for 24 h and examined for expression of CD86 (A) and MHCII (B) compared with an untreated rmGM-CSF only control (nil). Shown are means ± SEM from 4–5 independent experiments (**P < 0.01; *P < 0.05 referred control DC treated with LPS). (C) Mixed leukocyte reaction of infected DC and T cells. Cells were treated as described earlier and LPS-treated DC and non-treated DC (nil) were incubated with T cells for 5 d and examined for proliferation. Shown are the means ± SEM of three independent experiments.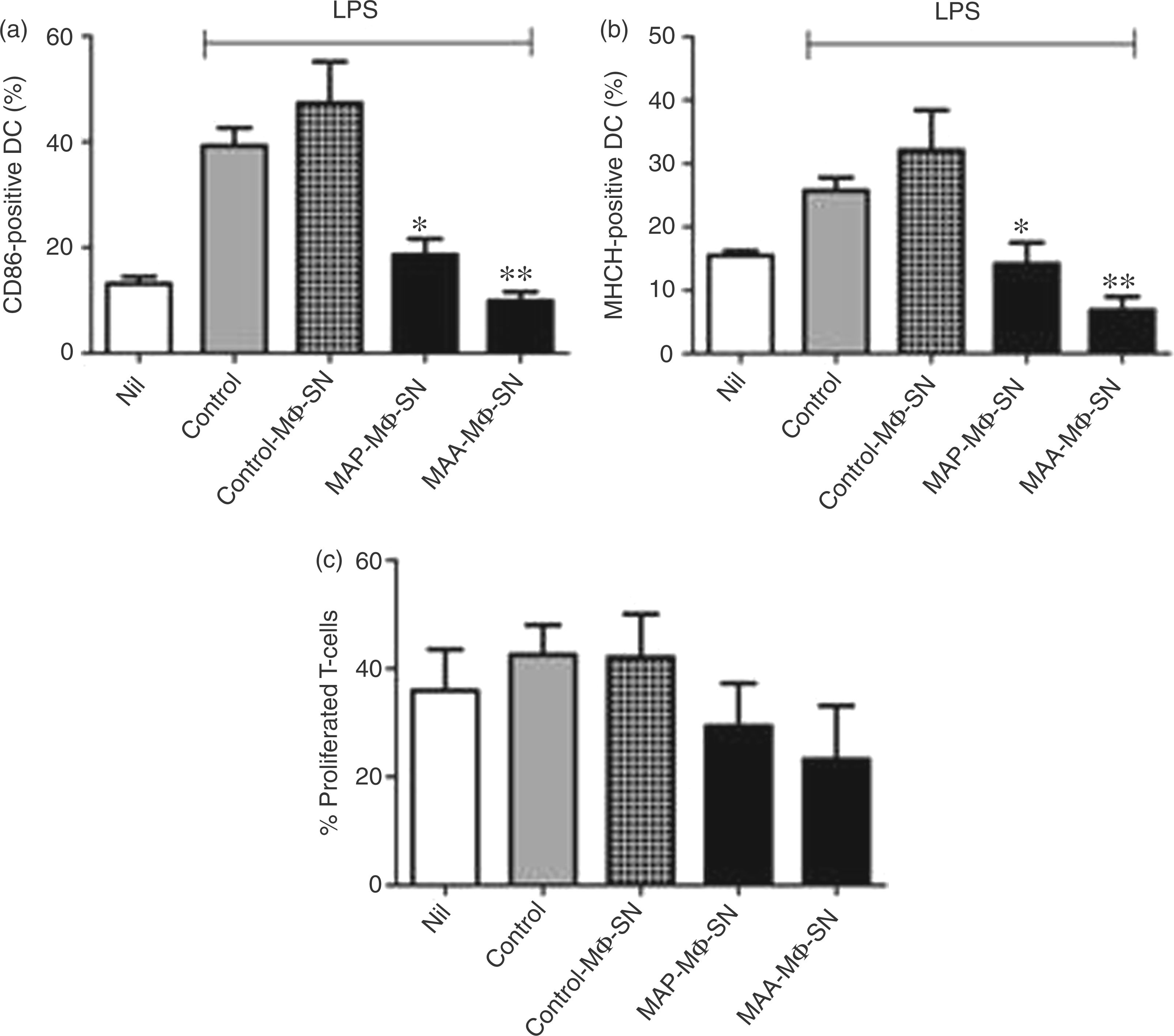
Next, we tested the capacity of DC incubated with MAP-MΦ-SN or MAA-MΦ-SN to induce proliferation of T cells by means of MLR. The results of three independent experiments are shown in Figure 3C. Even though we could not obtain data with statistical significance, the data clearly indicate a general tendency that the LPS-elicited ability of DC to cause T cell proliferation was impaired by treatment of DC with MAP-MΦ-SN or MAA-MΦ-SN, similar to DC without LPS stimulation (nil).
MAP- and MAA-infected macrophages are able to enhance IL-10, and subvert IL-12 and TNF-α release of maturation-impaired DC
DC were incubated with cell-free culture SN of MAP- and MAA-infected macrophages and treated with LPS as described earlier. The ability of DC to produce IL-10 and IL-12 was tested by ELISA of culture SN 24 h after LPS stimulation. As shown in Figure 4, LPS stimulation of non-treated cells or of cells treated with SN of non-infected macrophages induced secretion of IL-10 and IL-12. Incubation of DC with MAP-MΦ-SN and MAA-MΦ-SN resulted in significantly enhanced IL-10 production compared with the controls. In comparison with DC generated in the presence of control-MΦ-SN in MAP-MΦ-SN- and MAA-MΦ-SN-treated cells, IL-10 production was 2.9-fold and 2.6-fold higher respectively. Interestingly, the LPS-stimulation of control DC induced significantly higher IL-12 secretion than LPS-stimulation of DC generated in the presence of MAP-MΦ-SN and MAA-MΦ-SN. The release of IL-12 was 10.6-fold and 17.7-fold lower in MAP-MΦ-SN and MAA-MΦ-SN treated DC respectively. Also, the production of TNF-α was significantly lower after treatment with MAP-MΦ-SN and MAA-MΦ-SN.
IL-10, IL-12 and TNF-α production of SN-induced DC. Bone marrow-derived DC were cultured with SN of non-infected macrophages (control-MΦ-SN) or from macrophages infected with viable MAP- or MAA at MOI 10 for 24 h (MAP-MΦ-SN, MAA-MΦ-SN) and treated with LPS to induce cytokine release. Twenty-four h post-stimulation IL-10, IL-12 and TNF-α were measured in SN of DC (nil: non-treated DC; control: LPS-treated DC). Results are means ± SEM from 5–7 experiments (***P < 0.001; **P < 0.01; *P < 0.05; referred to DC treated with LPS).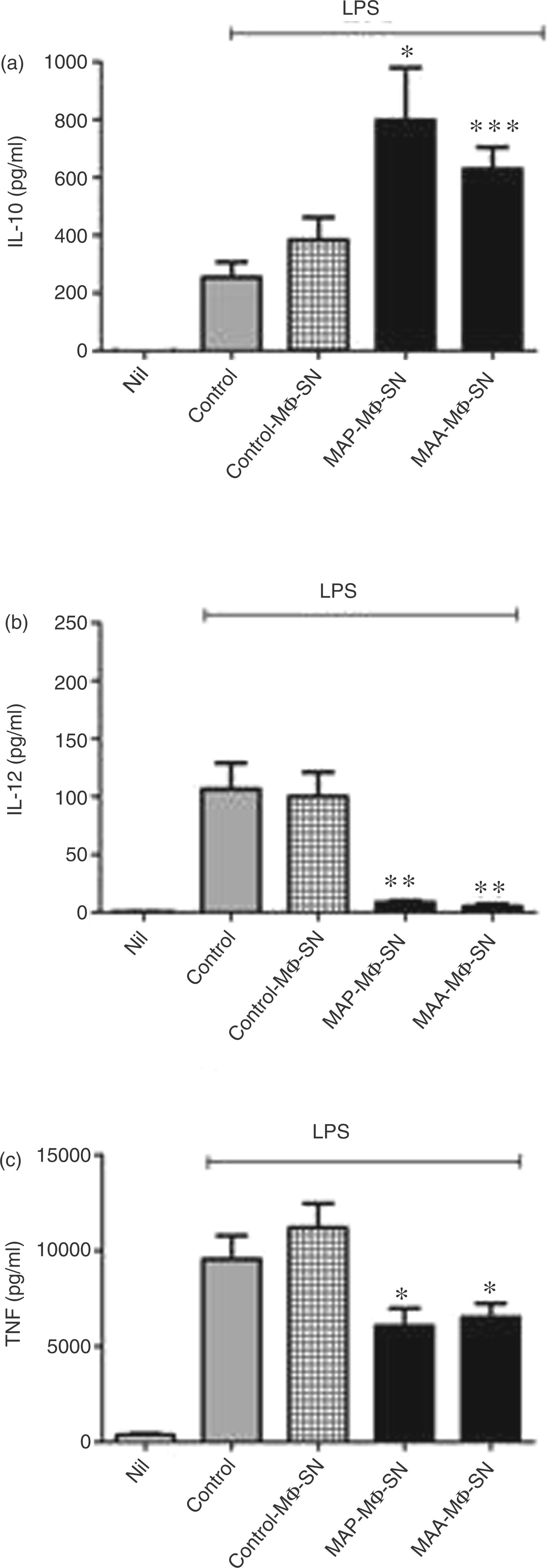
Characterization of MAP- and MAA-infected macrophage SN
We then analysed the SN for IL-6, IL-1β, IL-10, IL-12 and TNF-α production, cytokines that are known, or expected, to contribute to DC maturation.
23
As shown in Figure 5 A the pro-inflammatory cytokines IL-6 and TNF-α were detectable in high amounts, yet only weak IL-1β and IL-10, and no IL-12, levels were detected in the SN of infected macrophages. Persistence of mycobacteria in macrophages is associated with the release of mycobacterial components from the infected macrophage.
26
In agreement, as indicated by the clear signals in the immunoblot analyses of the SN from MAP-infected macrophages using an antiserum from a MAP-infected cow, we were able to detect antigenic mycobacterial components secreted from MAP-infected macrophages (Figure 5B). However, we were not able to identify the particular MAP components.
Characterization of the SN from MAP-infected macrophages. (A) J774.A1 macrophages were infected with live MAP at MOI 10 or left untreated (control). After 24 h SN were collected and assayed by ELISA for the production of IL-6, IL-1ß, TNF-α, IL-10 and IL-12. Means ± SEM from three experiments are shown. (B) SN concentrated by trichloroacetic acid precipitation from macrophages infected with viable MAP for 24 h (MAP-MΦ-SN) or untreated macrophages (control-MΦ-SN) were analysed by SDS-PAGE and immunoblotting with a serum of a MAP-infected cow. Whole cell lysate from MAP (wcl) served as a control for immunogenic mycobacterial components.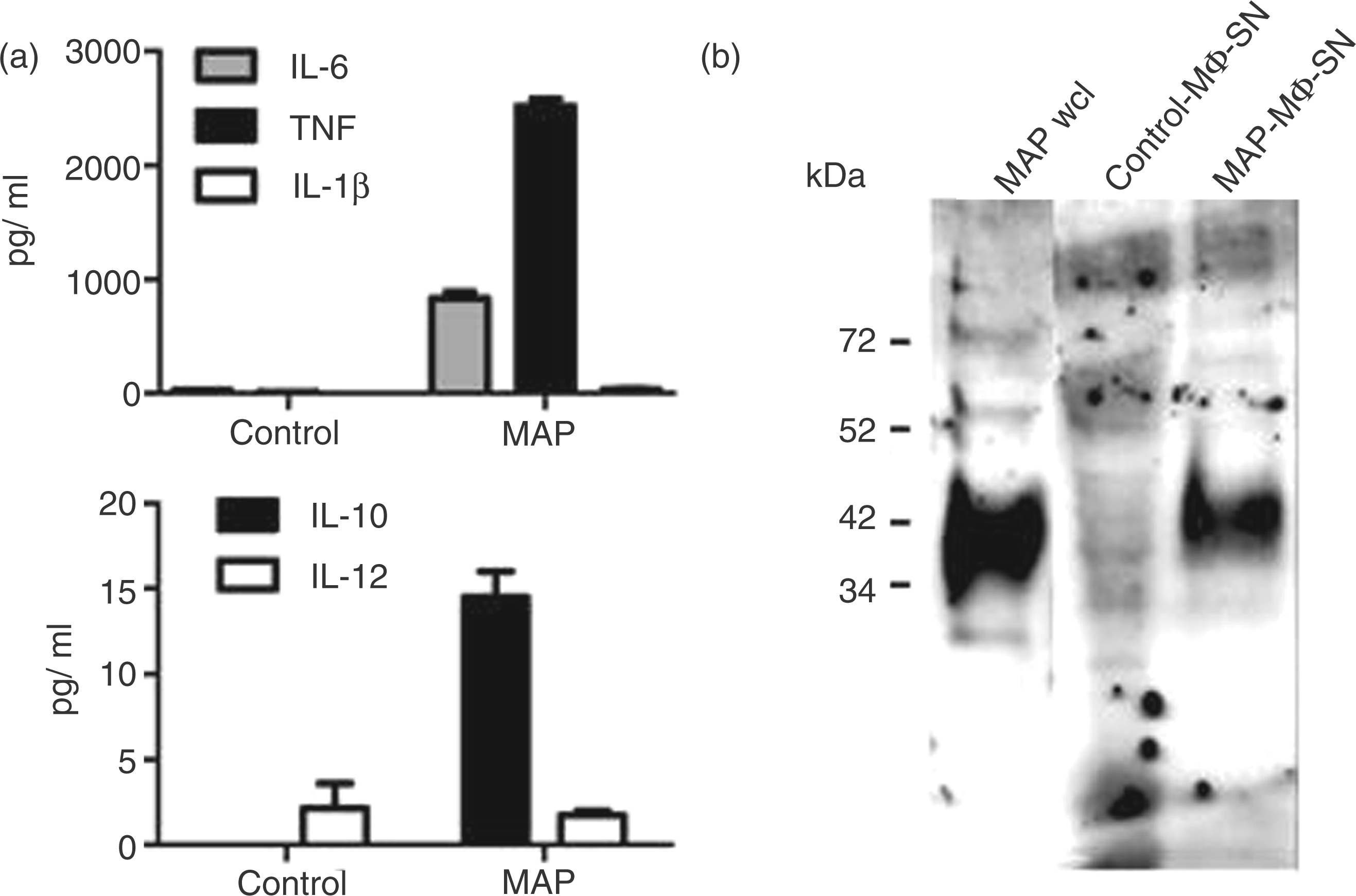
The role of cytokines in the process of DC maturation
IL-1β is known to induce DC to produce IL-12,27,28 whereas IL-6 plays a major role in maintaining immature DC.
29
In the macrophage SN we found high levels of IL-6, but only low levels of IL-1β. To define whether this cytokine profile might be relevant for the MAP-SN-induced DC phenotype, recombinant IL-6 and IL-1β were added to the cells during differentiation. As shown in Figure 6, the addition of IL-6 or IL-1β alone, but also together with IL-1β, was able to lower expression of CD86 and MHCII following LPS treatment. Furthermore, IL-6 treatment reduced LPS-induced IL-12 release, but did not influence LPS-induced IL-10 release. However, the combined treatment of the cells with IL-6 and IL-1β reduced LPS-induced IL-12 release and enhanced LPS-induced IL-10 release. This is in contrast to the function of IL-10, which leads to a high expression of IL-12 and has no effect on the expression of MHCII and CD86.
DC maturation after incubation with IL-6 and IL-1β. Bone marrow-derived DC were cultured in complete medium supplemented with GM-CSF and treated at d 3, 6 and 8 with cell-free SN from non-infected macrophages (control-MΦ-SN) alone, or with control-MΦ-SN supplemented with IL-6, IL-1β, and IL-6 and IL-1β (10 ng/ml each). At d 9 DC were stimulated with 0.375 µg/ml LPS (control) for 24 h or left untreated (nil) and examined for expression of CD86 and MHCII (A) and for expression of IL-10 and IL-12 by ELISA (B). Shown are means ± SEM from 4–5 independent experiments (**P < 0.01; *P < 0.05 referred control DC treated with LPS).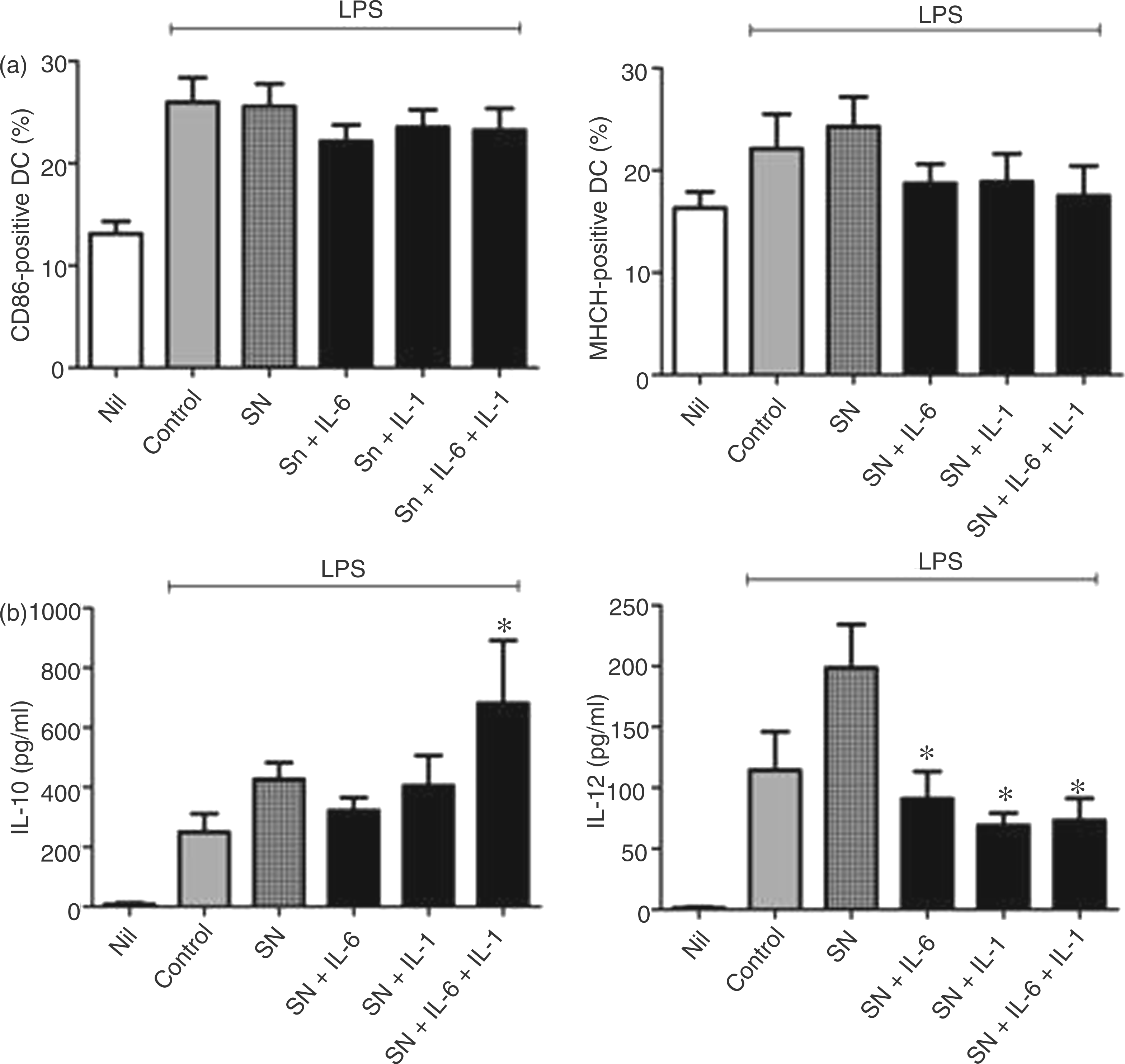
Discussion
In the present study we were interested in the interaction of MAP and MAA with DC. We demonstrated that MAP and MAA are able to manipulate maturation and function of DC in two ways: (i) directly by infection and persistence in the DC and (ii) indirectly via the milieu created by secreted components of the mycobacteria-infected macrophage. DC responses were considerably subverted to an extent that might result in insufficient initiation of a Th1-based immune reaction.
It is known that all M. avium ssp. infect their hosts via enterocytes or M-cells, and will be recognized by subepithelial macrophages, as well as intestinal DC. Even though macrophages are clearly defined as target cells for MAP and MAA persistence, the role of DC is still unclear. Among the M. avium ssp. only MAP causes severe intestinal disease and one might argue that this is based on DC dysfunction caused by MAP.
DC derived from murine bone marrow on the basis of the protocol of Lutz et al. 23 represent a standardized population that has been used successfully in numerous studies on the interaction of DC with different pathogens,30,31 as well as for analyses of a putative virulence protein of MAP. 32 The protocol is based on the generation of DC from murine bone marrow precursor cells in the presence of rmGM-CSF. In this way, the differentiation towards CD11c+ DC precursor cells, already expressing low levels of CD86 and MHCII, occurs. Final maturation of these cells is obtained by a bacterial stimulus, such as LPS and is indicated by the expression of enhanced levels CD86 and MHCII, as well as an enhanced capacity to activate T cells.
We used the above protocol to analyse the effects of MAA and MAP on DC maturation. In this study we found that maturation of DC by MAP and MAA was similar to the maturation following LPS stimulation, as far as MHCII and CD86 expression is concerned. This effect was independent of bacterial viability. Accordingly, mycobacteria-maturated DC were able to induce T cell proliferation in a mixed leukocyte reaction. We also tested the stimulatory capacity of viable and heat-killed MAP for IL-10 and IL-12 production. Interestingly, there were significant differences in the cytokine profiles comparing LPS stimulated DC with MAP- or MAA-infected DC. While the amount of IL-10 in SN of mycobacteria-infected DC was higher than that in LPS-treated DC, the IL-12 secretion from infected DC was considerably lower than that of LPS-matured DC. This indicates that mycobacteria-infected DC maturate in a similar way as LPS stimulated cells as far as MHCII and CD86 is concerned. However, in contrast to maturation induced by LPS, mycobacteria-induced maturation is accompanied by a strong anti-inflammatory response. Viable and heat-inactivated bacteria behaved similar in both types of experiments with limited differences indicating that heat-stable structural components of MAP and MAA are the major contributors for MAP- and MAA-induced DC maturation.
DC maturation is influenced mainly by microbes and microbial components and may be further affected by cytokines from non-infected and infected bystander cells, such as lymphocytes or macrophages. Thus, Rimoldi et al. 33 described an Ag-independent activation of DC by the release of mediators from neighbouring epithelial cells infected with Salmonella Typhimurium. As macrophages represent the primary host cell for mycobacterial persistence, 34 we also tested whether mycobacteria-infected macrophages modulate DC maturation. Interestingly, SN of MAP- or MAA-infected macrophages inhibited the LPS-induced final maturation of DC and induced a semi-mature, tolerogenic DC. Most interestingly, these immature CD11c+ cells displayed significantly lower IL-12 production and an increase in IL-10 production. This indicates that DC maturation might be influenced through the infected macrophages. However, already at the level of the DC precursor, suppression of the pro-inflammatory response necessary for a Th1 directed T cell response occurs.
We found that the SN-induced DC phenotype was reversible if heat- or trypsin-treated SN were used (data not shown), indicating that proteinous factors might be involved. Their origin might be secreted macrophage factors, such as cytokines, or mycobacterial components released from the macrophage during infection, or both.
The significance of cytokines for DC maturation is emphasized by the fact that single cytokines, such as IL-10, IL-1β, or IL-6, have considerable effects on DC. Thus, IL-10 is known to affect the maturation of DC in vitro.35,36 IL-1β is known to induce DC to produce IL-12,27,28 whereas IL-6 plays a major role in maintaining immature DC. 29
In a more recent study Remoli et al. 37 reported that inhibition of DC differentiation by SN from M. tuberculosis macrophages resulted from induced IL-10. Our data show that the DC phenotype presented here is IL-10 independent. Thus, MAP-infected macrophages produced only low amounts of IL-10 after 24 h of infection. Furthermore, in contrast to the results of Remoli et al., 37 we tested the final maturation capability of DC generated in the presence of SN from MAP- or MAA-infected macrophages by stimulating them with LPS. Under these conditions the addition of recombinant IL-10 in macrophage medium during DC maturation had no additional effect (data not shown). In fact, our SN-induced DC phenotype resembled that of tolerogenic DC, which is suggested to be induced by IL-6 and/or by stimulation with TLR ligands at low concentrations. 38 Moreover, our macrophage SN exhibited high levels of IL-6, but only low levels of IL-1β. To prove the hypothesis that this feature of the SN might contribute to the SN-induced DC phenotype under our conditions, we tested effects of recombinant IL-6 on the DC maturation process. Our experiments support that IL-6 might contribute to the SN-induced DC phenotype, as it was able to reduce CD86, MHCII and IL-12 expression. However, IL-6 alone was unable to induce the complete SN-induced phenotype indicated by the missing IL-10 induction. Indeed, the induction of IL-10 by IL-6 and IL-1 β indicates that DC, in addition to IL-6, need other pro-inflammatory stimuli for IL-10 production.
We have shown previously that MAP and MAA infect murine macrophage cell lines, and that the infection is accompanied by release of cytokines and mycobacterial persistence for long time periods without affecting the cellular integrity.3,22 In this study we were able to identify mycobacterial Ags in the SN of MAP infected macrophages. Most probably, these components were released from the macrophage by the formation of exosomes containing mycobacterial material. 39 Exosomes from M. tuberculosis-infected macrophages have been shown to contain mycobacterial components, including lipoarabinomannan and other mycobacterial lipids. 39 Furthermore, exosomes from M. tuberculosis and M. bovis BCG-infected macrophages were found to activate immune cells in vivo and in vitro.26,40 As lipoarabinomannan and lipomannan were recognized by the antiserum from paratuberculosis diseased cows (data not shown) it seems that, beside proteins, mycobacterial lipids were also present in the SN from mycobacteria-infected macrophages. Thus, in our system these components may provide the pro-inflammatory stimulus needed for IL-10 production. From this we assume that the semi-mature DC phenotype results from combined effects of secreted cytokines and released antigenic mycobacterial components of the infected macrophage. Nevertheless, we cannot exclude that other, or additional, factors may influence DC maturation. This, and the precise role of IL-6, as well as the relevance of the mycobacterial components, awaits further study.
Both approaches on mycobacterial effects on DC described in the present study point out clearly that MAP and MAA manipulate the DC response for subverting the anti-mycobacterial immune response. Thus, the mycobacterial effects on DC resulted in high production of anti-inflammatory IL-10 and low production of IL-12, which is advantageous for mycobacterial persistence as IL-12-directed Th1 polarization will be influenced. Furthermore, increased IL-10 production might play an important role in maintaining MAP and MAA infections, as it may account for the survival of mycobacteria through its inhibitory activity on anti-mycobacterial macrophage functions.41,42 We found similar effects on DC by MAP and MAA. Nevertheless, our findings might be of particular importance for MAP infections as an increased expression of IL-10 mRNA was found in the affected intestinal tissues and lymph nodes of cows with paratuberculosis.43–45 The source of IL-10 in the tissues, however, is still unknown.
In summary, our findings demonstrate that MAP and MAA may subvert DC effector functions directly by the infection of the DC and indirectly via the milieu created by secretory components of the mycobacteria-infected macrophage. In both ways MAP and MAA may considerably sustain their own persistence and the progression of disease.
Footnotes
Acknowledgements
We are grateful to Viktoria Gaarder and Sabine Goebel for their excellent technical assistance, and to Joerg Merkel for excellent computer technical support.
Funding
This work was supported by the Germany Ministry for Science and Education (BMBF, 01KI0750 & 01KI1003A). R.G. was additionally supported by a grant from the German Research Foundation (DFG, Go983/1).