
Abstract
Infectious diseases have been postulated to play an important role in exerting pressure and in selection of TLR polymorphisms. Single nucelotide polymorphisms (SNPs) of TLR4 have been reported to show unique distributions in populations from Africa, Asia and Europe, and malaria is suggested to influence these patterns. In this context, we examined association of TLR polymorphisms with the risk of malaria in two ethnic groups—the Austro-Asiatics and Tibeto-Burmans—from malaria endemic districts of Assam to understand the influence of malaria in selection of TLRs in these genetically-distinct populations. TLR9 (T-1237C) mutation was positively associated with complicated (P = 0.001) and frequent (P = 0.035) malaria in Austro-Asiatics (relative risk = 0.595 95% CI: 0.479–0.836), but not in Tibeto-Burmans. Nonetheless, these alleles were not in Hardy-Weinberg Equilibrium in Tibeto-Burmans (P < 0.001). In contrast, the TLR9 1486T/C genotype was favourable where it was negatively associated with complicated malaria (Fishers exact P = 0.014). Sequencing data revealed that the two populations differed in nucleotide diversity of the TLR9 promoter region. Enhanced expression of TLR4 (P = 0.05), but not of TLR9, was associated with complicated malaria. Austro-Asiatics appeared to have accumulated favourable genotypes of TLR9, perhaps because of their longer exposure to malaria.
Introduction
Innate immunity is gaining importance in the understanding of immune responses to infectious organisms. Cells of the immune system armed with germ-line encoded receptors (pathogen recognition receptors) of limited repertoires respond to the molecular motifs on exogenous antigens by secreting cytokines and chemokines that significantly influence adaptive immunity and disease outcome. 1
TLRs are an important component of the innate immune system. Their role in bacterial and viral infections is well documented and they are becoming important in immune responses to protozoan infections.2–4 Activation of TLRs leads not only to the robust production of pro-inflammatory mediators, but also to the production of unique effectors, which provide pathogen-tailored immune responses. In malaria, Plasmodium falciparum glycosylphosphatidylinositol (GPI) has been shown to induce the expression of proinflammatory cytokines and immune mediators in vitro. 5 Furthermore, GPI was reported to induce signalling via both TLR2 and TLR4, whereas haemozoin (HZ), in combination with plasmodial DNA, activated dendritic cells by engaging TLR9. 6 Changes in TLR responses also modulated the production of inflammatory cytokines and thus prevented the development of severe pathology, emphasising the role of these responses in malarial pathogenesis. 7 Increased expression of TLR genes was also seen in P. falciparum infected subjects and it was associated with enhanced IFN-γ, TNF-α and IL-10 production. 8
Genetic variations of TLRs can change the ability of the organism to respond to different stimuli, hence the interest in the possible associations of TLR polymorphisms with altered susceptibility to infections and inflammatory diseases is growing. 2 Studies have linked TLR2 (Arg753Gln) with rheumatic fever in children and TLR4 polymorphisms with increased susceptibility to bacterial infections.9–11 TLR9 promoter polymorphisms (T-1237C and T-1486C) have been assumed to influence transcription regulation and they have been associated with asthma, Crohns disease, atopic eczema and lymphoma.12–14 Many studies have also implicated polymorphisms of TLR2, 4 and 9 in the disease manifestation of malaria. Single nucleotide polymorphisms (SNPs) of TLR4 (Asp299Gly) and TLR9 (T-1486C), but not TLR9 (T-1237C), were associated with low birth rate, maternal anaemia and with high parasitaemia, suggesting that these SNPs play a role in the manifestation of malaria during pregnancy.15,16 SNPs of TLR4 Asp299Gly and Thr399Ile were seen to increase the risk of severe malaria in Ghanian children.15,17
A number of SNPs associated with inflammatory and immunologic diseases also show large frequency differences among ethnically-distinct populations.2,18 A study in India, showed that high frequency of FcγR2A exon4 AA genotype protected the Tharus, a population living in the foothills of the Himalayas, from falciparum malaria. 19
Distribution of TLR polymorphisms have been shown to differ among ethnically different populations in Asia, Africa and Europe, and malaria was suggested to be the driving force. 20 In this context, we have examined the association of SNPs in TLR4 and 9, and the expression of these genes in relation to malaria and ethnicity in Assam State of northeast India, in which malaria is endemic. 21 We report the differential association of polymorphisms and expression of TLR9 with malaria in two ethnic groups. Increased expression of TLR4 gene was seen in complicated malaria. The Austro-Asiatics (plains) had the favourable genotype, conferring protection from complicated malaria.
Materials and methods
Study sites
The study was conducted at two study sites: Guabari, a village of Baksa district which lies at the foothills of Bhutan; and at Kondoli in Karbi Anglong foothills of Nagaon district of Assam. The two study sites may be classified as mesoendemic for malaria. The characteristics of the two study sites have been described previously. 22
Study participants
Patients were enrolled into the study after obtaining written informed consent. Active, as well as passive, case detection was followed which was carried out by local health workers in Guabari, while at Kondoli it was done by hospital staff. Based on linguistic group affinities, the study population was stratified into two groups, namely the speakers of Tibeto-Burman (TB) that included the Bodo-Kachari and Nepalis, the Austro-Asiatic (AA) that comprised the tea tribes who are Mundari speakers. 19 Sampling was done such that the samples were drawn independently and randomly from each group, particularly at Guabari, which has an ethnically-mixed population. Though the people belonging to AA and TB groups are found in Assam at present, but their origin is very different. The AA group is believed to include the earliest inhabitants of Indian plains whereas the TBs are believed to have migrated from South China and Tibet approximately 5000–10,000 years ago.23–25 Moreover, based on mitochondrial DNA and NRY (non-recombing region of the Y chromosome) haplotypes, the AA was found to be genetically distinct from TB. 25 Khasis are a group of people residing in the state of Meghalaya and they are AA because their language belongs to the Khasi-Khmic subfamily of the AAs. 26,27 In the present study, the AA group from Assam was denoted as AA (plains) and the Khasis as AA (hills). Khasi participants (n = 30) were from hilly regions (>4900 ft above sea-level) where exposure to malaria is unlikely and so served as controls for AA (plains) in SNP-malaria association studies.
Sample collection
Blood samples were obtained from individuals, irrespective of their P. falciparum-positive (Pf+) status at the time of collection. Inclusion criteria were individuals with Pf+ or with history of falciparum malaria. Exclusion criteria were children with age less than 1 year, pregnant women and individuals suffering from any other diseases. Plasmodium falciparum positivity was checked using Rapid Diagnostic Kits (FalciVax; Orchid Biomedical Laboratories, Goa, India), and confirmed by microscopy of thin and thick blood smears. Individuals were categorized according to their disease symptoms and frequency of malaria incidence, which were recorded simultaneously. Complicated malaria was defined following World Health Organization (WHO) guidelines. 28 Uncomplicated malaria included fever, headache, body ache, and other mild symptoms or asymptomatic cases with parasitaemia of ≤5000/µl of blood. On the basis of number of clinical episodes of malaria, the participants were classified as infrequent (≤2 episodes) or frequent malaria (>2 episodes) groups as detailed in our earlier work. 29 The study was approved by the Tezpur University Ethical Committee (Resolution number 3 dated 13 June 2006).
SNP typing of TLR4 and 9
Two hundred microlitres of blood were collected in EDTA-tubes and genomic DNA extraction was carried out using Qiagen blood extraction kit (Qiagen, Hilden, Germany). The TLR4 and 9 genes were amplified by PCR and then digested using restriction enzymes, as described.10,30–32 The fragments were then analysed on 3% agarose gels pre-stained with ethidium bromide.
Sequence analysis
TLR9 promoter regions were sequenced (Sequencer; Applied Biosystems, Foster City, CA) and aligned with the sequence available in the GenBank (Accession number: NT_022517.18) using CLUSTAL X and the matrix selected for the purpose is Clustal W 1.6. The alignment file created was used for further analysis using DNASp V 5.0. TLR9 promoter sequences have been submitted to GenBank with the following accession numbers: HM231310, HM231311, HM231312 and HM231313.
TLR gene expression study
The cohort used for the TLR4 and 9 genotyping study was also used for expression study. Forty-six samples were drawn randomly from each of the three diseased groups (comprising of patients having complicated malaria, uncomplicated malaria and non-malarial fever) and from the healthy control group. Total RNA was then extracted from these blood samples and stored in RNA later, as per the manufacturer’s instructions (Ribopure; Ambion, Invitrogen BioServices, India). Reverse transcription (RT) reactions were set up by using cDNA Archive kit (Applied Biosystems, Foster City, CA). Real time PCR was performed in 7300 Real time PCR system (Applied Biosystems, Foster City, CA). TLR genes expression was calculated in relation to the expression of RnaseP, which was used as an endogenous control. 33
Data analysis
The data was analysed using Excel Stat Software, 2010 version. Variation in the frequency of the TLR4 and 9 genes with respect to disease status, frequency of disease and ethnicity was analysed by Fisher’s exact test. The allele frequencies were calculated (estimation of P values by Pearson test) to test if they were in agreement with Hardy–Weinberg equilibrium (HWE). For TLR gene expression data analysis we used the comparative CT method (ΔΔCT method). Expression of TLR genes was analysed by t-test used for comparing the means in the different groups. Expression levels of TLR genes were compared between the younger age group (≤10 years) and older age group (>10 years) who had malaria at that point in time.
Results
Overall frequency of SNPs of TLR genes
Comparison of the genotype frequencies of the TLR4 and 9 loci in the study population. Analysis was performed to check the interactions in the three genotypes of each TLR gene by Fisher’s exact test.
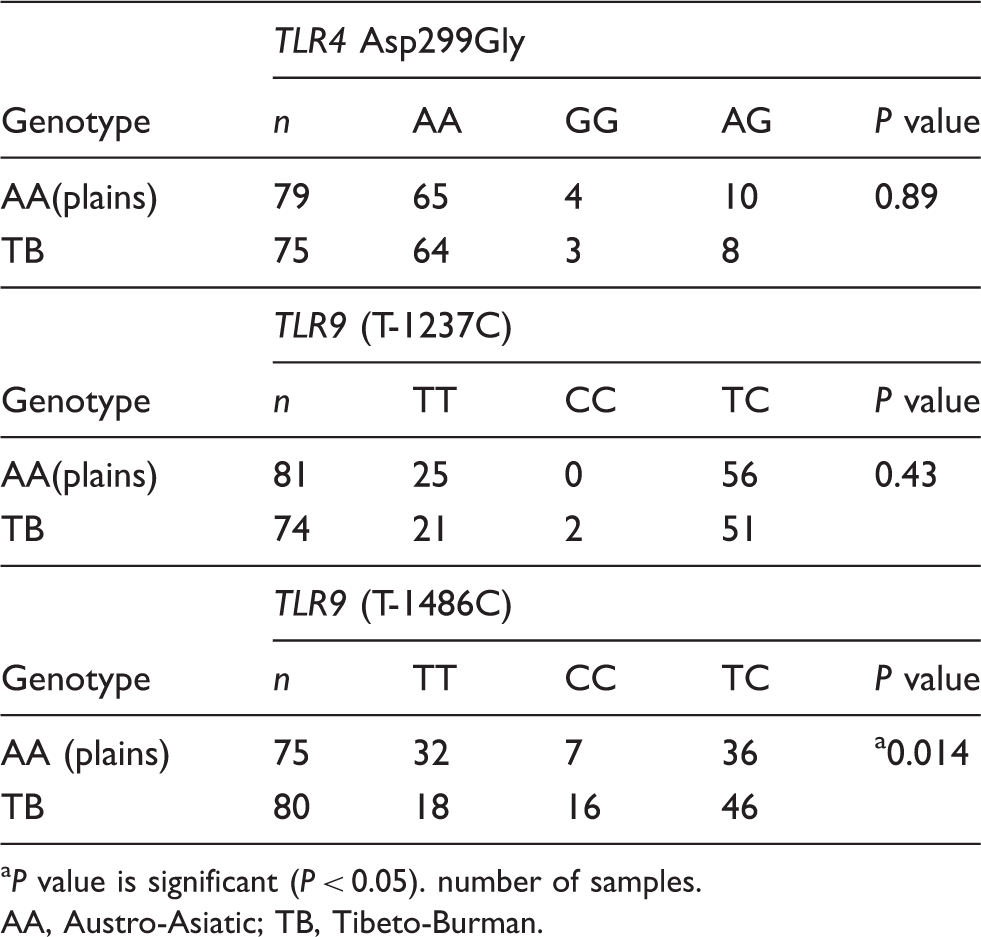
P value is significant (P < 0.05). number of samples.
AA, Austro-Asiatic; TB, Tibeto-Burman.
Comparison of the allele frequencies of the SNPs of TLR4 and 9 loci in the study population.
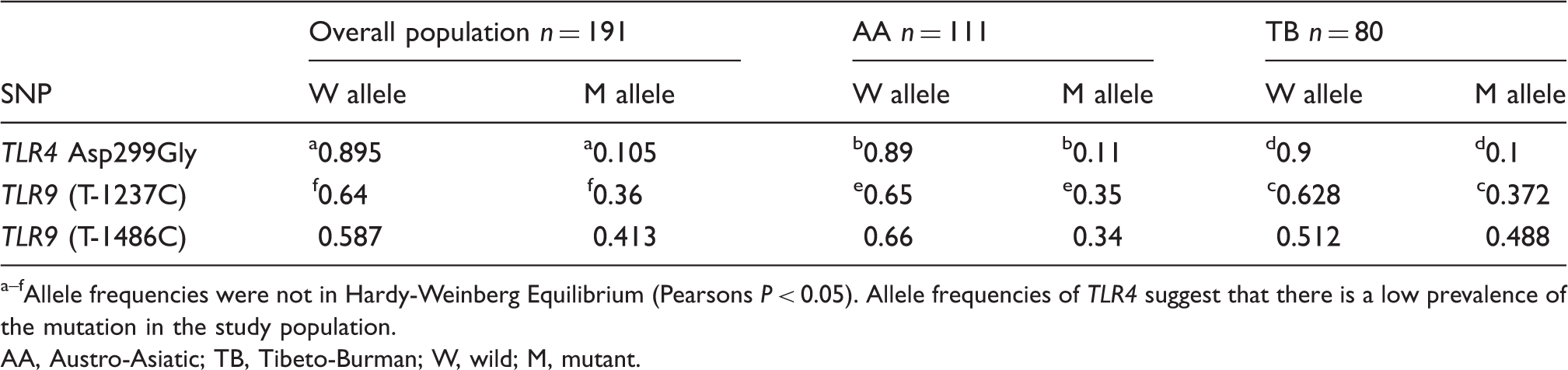
Allele frequencies were not in Hardy-Weinberg Equilibrium (Pearsons P < 0.05). Allele frequencies of TLR4 suggest that there is a low prevalence of the mutation in the study population.
AA, Austro-Asiatic; TB, Tibeto-Burman; W, wild; M, mutant.
Comparison of the genotype frequencies of the TLR4 and 9 between Austro-Asiatics (AA) (plains) and AA (hills).
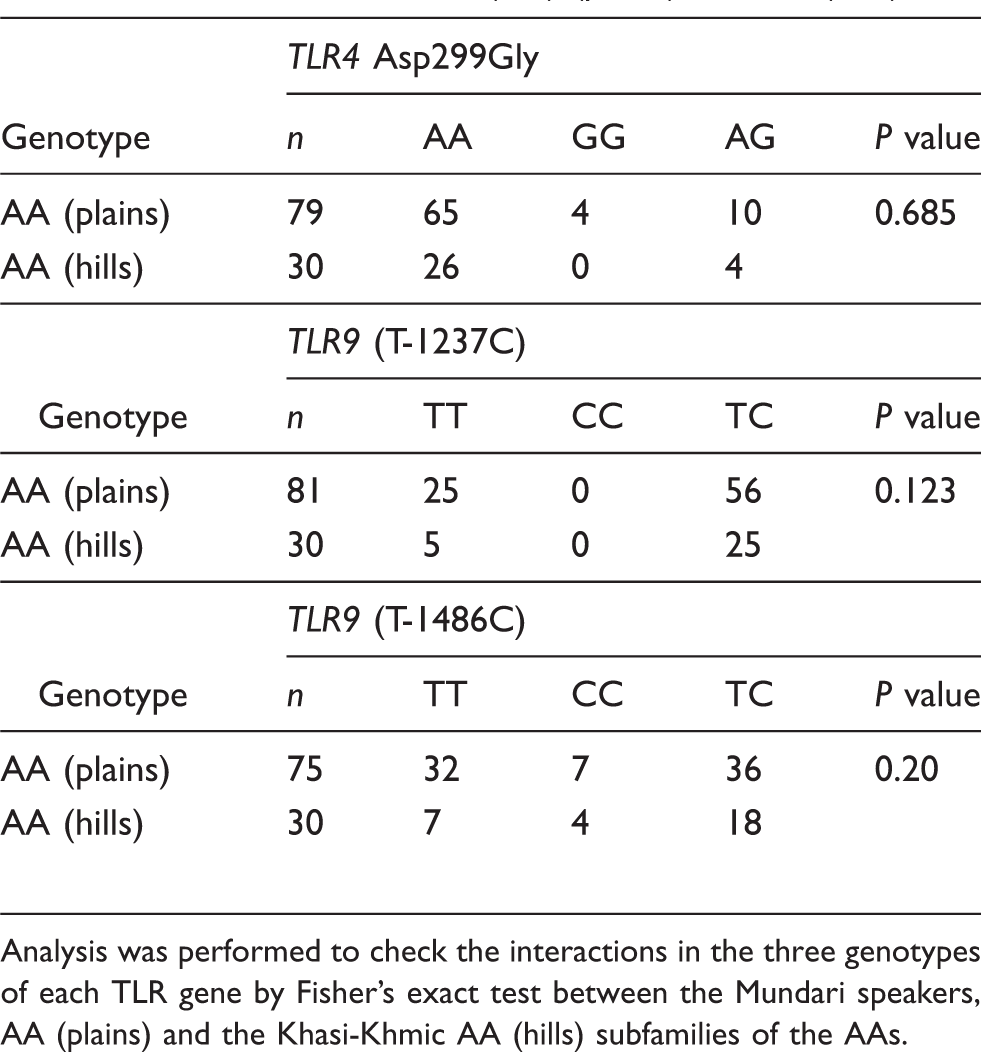
Analysis was performed to check the interactions in the three genotypes of each TLR gene by Fisher’s exact test between the Mundari speakers, AA (plains) and the Khasi-Khmic AA (hills) subfamilies of the AAs.
Sequence analysis
Differences in SNP frequencies in the two ethnic groups were further analysed by sequencing the PCR products of TLR9 promoter region. The sequences were compared with the sequence data present in Gen Bank (GenBank accession number: NT_022517.18). Analysis of the two subject populations with the sequence present in Gen Bank showed that the TB population had a maximum variation in the nucleotide sequence in the range of nucleotide from 162–340 over 178 base pair (bp) length (Figure 1A). In contrast, in the AA population, variation was seen in the later part of the sequence over a region of 180 bp, lying in range of nucleotide number 420–600 (Figure 1B).
Comparison of nucleotide divergence. The figure compares nucleotide divergence (A) between Tibeto-Burman (TB) and GenBank (NT_022517.18) (B) between Austro-Asiatic (AA) and GenBank (NT_022517.18) and (C) between AA and TB. Pi, nucleotide diversity, the average number of nucleotide differences per site between two sequences; Dxy, average number of nucleotide substitution per site between populations; Da, number of net nucleotide substitutions per site between populations. Alignment of the sequences was done using CLUSTAL X and further analysis using DNASp V 5.0.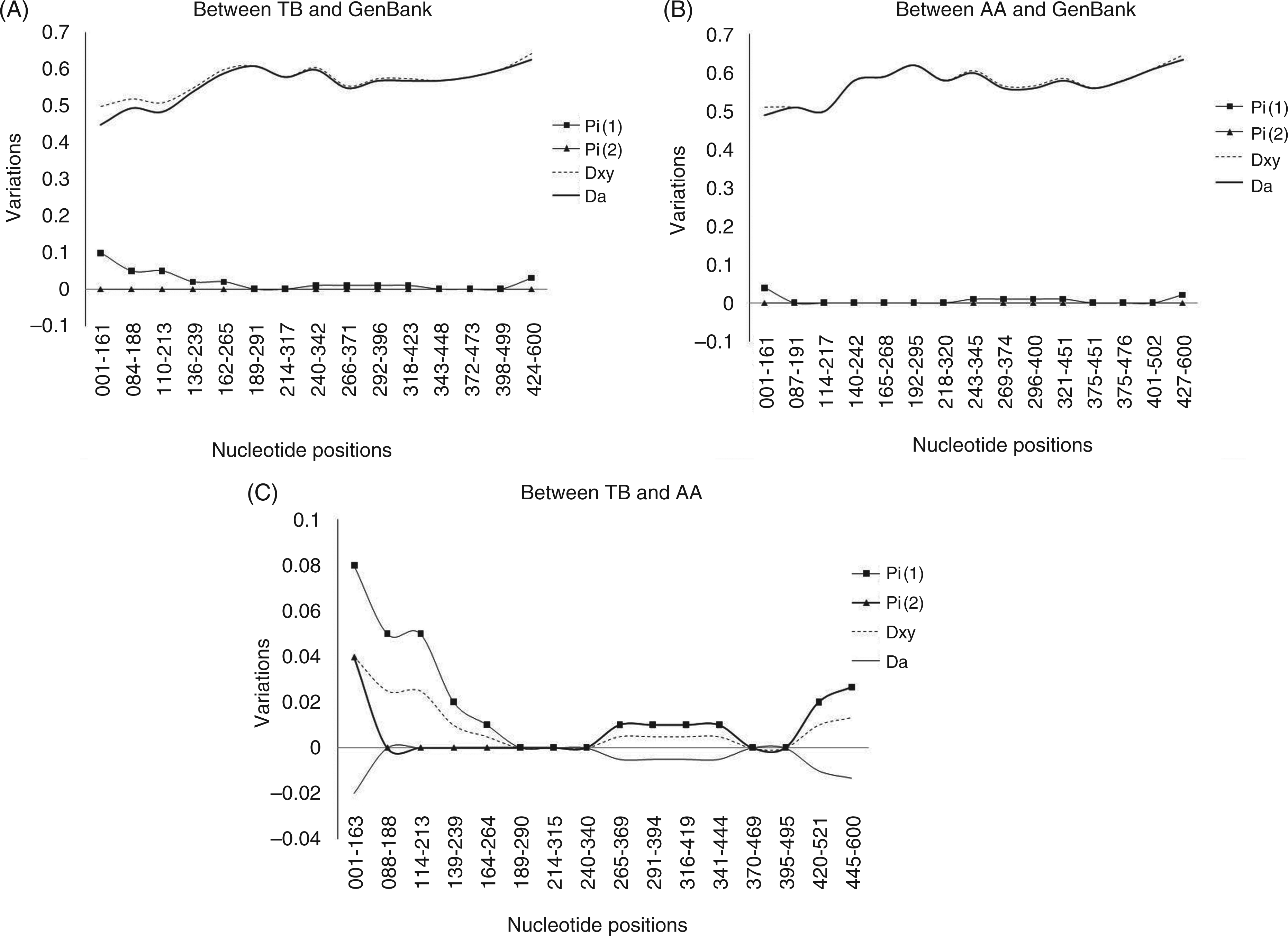
Analysis between the two populations showed a distinct pattern in the number of nucleotide variation, as shown in Figure 1C, where the TB population showed maximum variation in the first 200 bp region while the AA population was seen to show maximum variations in the 400–600 bp region.
Association of SNPs of TLR4 and 9 genes with malaria
We analysed the frequency of SNPs in relation to disease severity: complicated malaria and uncomplicated malaria and with respect to frequency of episodes.
Complicated and uncomplicated malaria
Comparison of the genotype frequencies of the SNPs of TLR4 and 9 in the two ethnicities in complicated and uncomplicated malaria groups. Analysis was performed to check the interactions in the three genotypes of each TLR gene.
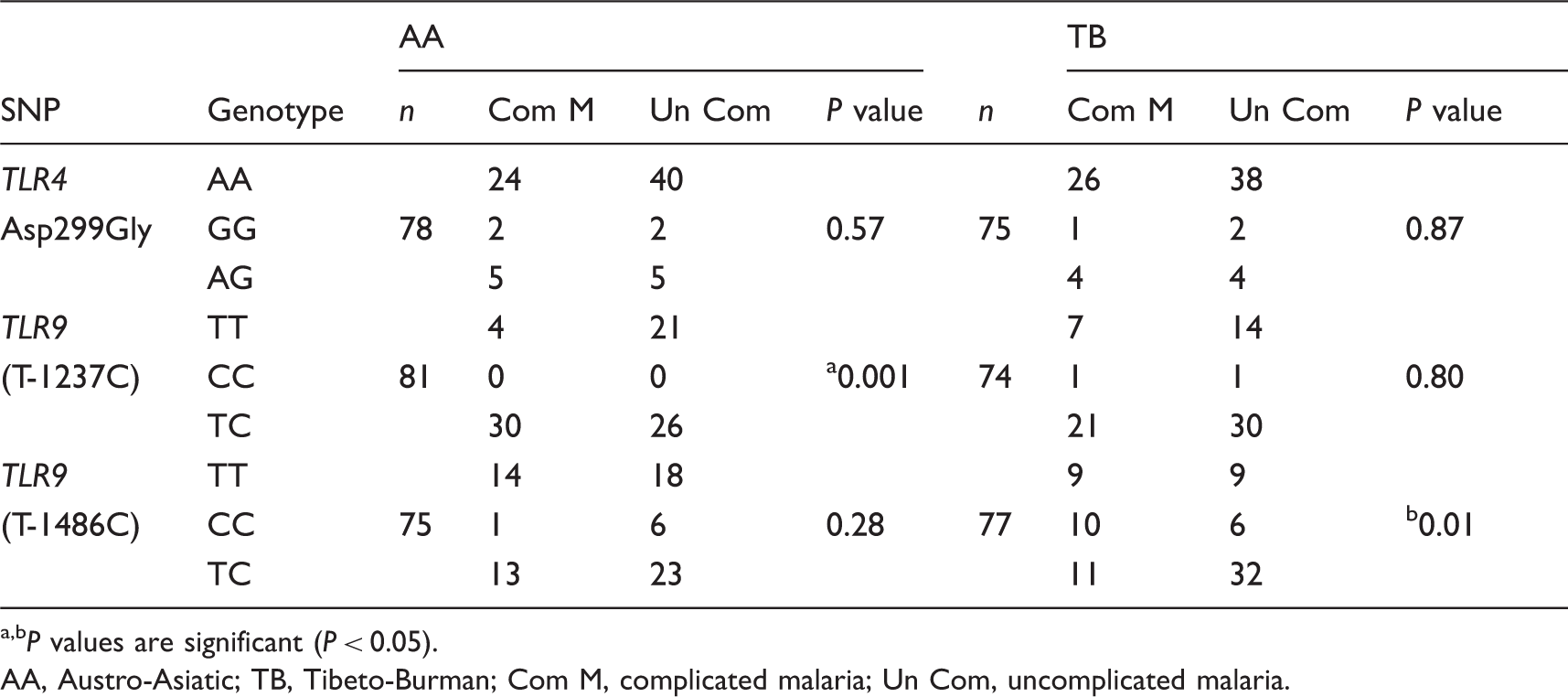
P values are significant (P < 0.05).
AA, Austro-Asiatic; TB, Tibeto-Burman; Com M, complicated malaria; Un Com, uncomplicated malaria.
Frequency of the disease (number of episodes)
Comparison of the genotype frequencies of the SNPs of TLR4 and TLR9 in the two ethnicities in frequent and infrequent malaria episodes.
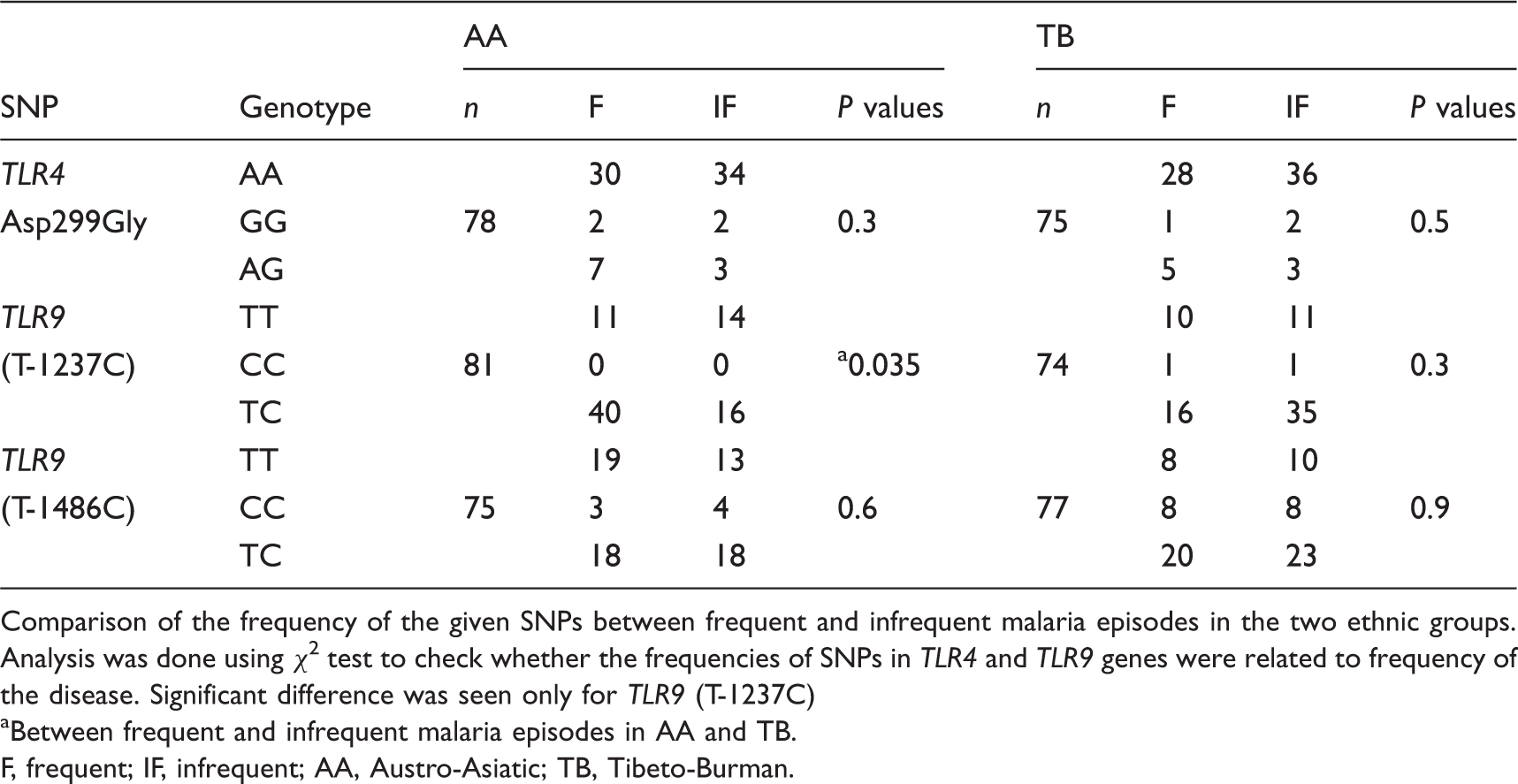
Comparison of the frequency of the given SNPs between frequent and infrequent malaria episodes in the two ethnic groups. Analysis was done using χ2 test to check whether the frequencies of SNPs in TLR4 and TLR9 genes were related to frequency of the disease. Significant difference was seen only for TLR9 (T-1237C)
Between frequent and infrequent malaria episodes in AA and TB.
F, frequent; IF, infrequent; AA, Austro-Asiatic; TB, Tibeto-Burman.
SNP grouping of TLR9
SNP-grouping of TLR9 (T-1237C and T-1486C) in the study population. Black and white fields denote the presence and absence of wild or mutant alleles respectively.
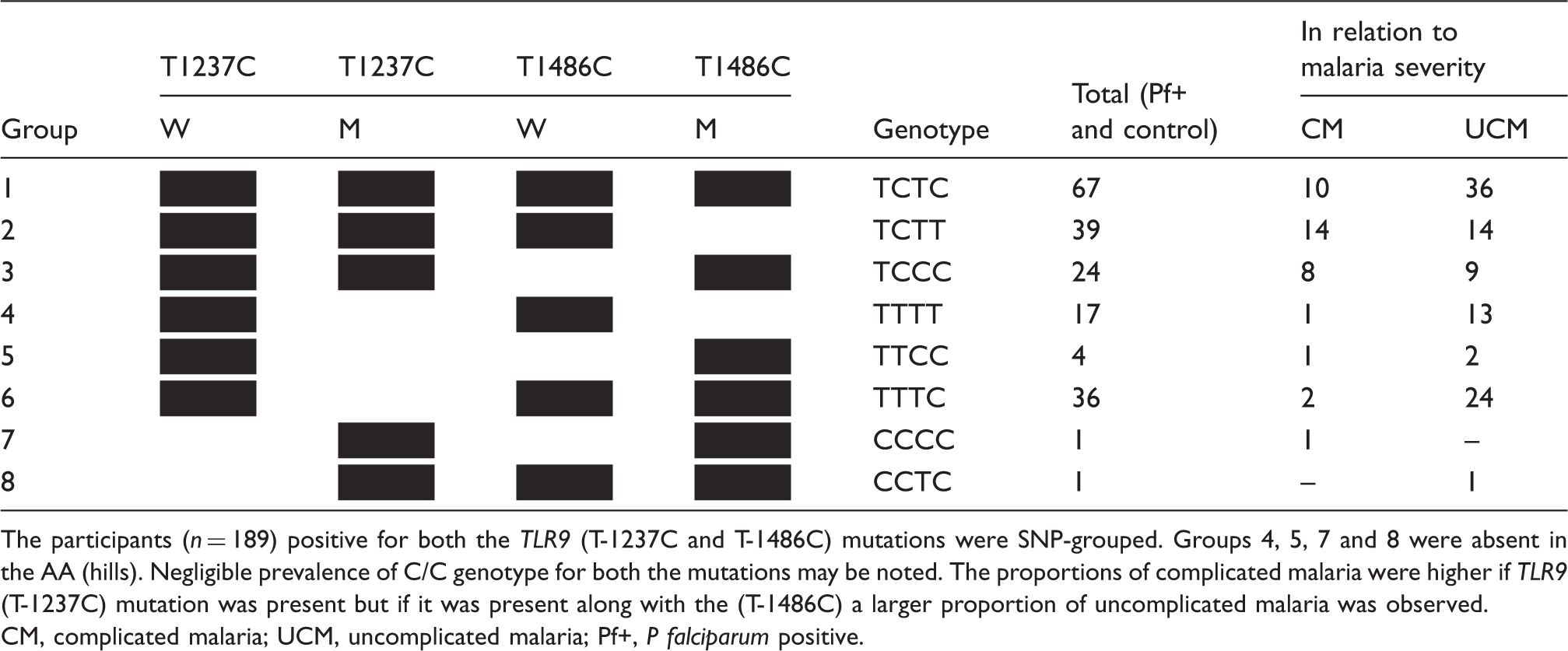
The participants (n = 189) positive for both the TLR9 (T-1237C and T-1486C) mutations were SNP-grouped. Groups 4, 5, 7 and 8 were absent in the AA (hills). Negligible prevalence of C/C genotype for both the mutations may be noted. The proportions of complicated malaria were higher if TLR9 (T-1237C) mutation was present but if it was present along with the (T-1486C) a larger proportion of uncomplicated malaria was observed.
CM, complicated malaria; UCM, uncomplicated malaria; Pf+, P falciparum positive.
TLR4 and 9 genes expression
Forty-six samples were analysed for the mRNA levels of TLR4 and 9 genes and data analysed in relation to disease status. TLR4 and 9 mRNA levels showed no difference between malaria and no malaria groups (P > 0.05). When the data were analysed in the context of complicated and uncomplicated malaria (Figure 2A), TLR4 gene expression was higher in the complicated group (P = 0.05), whereas in the uncomplicated group it was lower in comparison with the no malaria group.
Comparison of expression of TLR4 and TLR9 genes with disease severity and age. Mean expression of TLR4 and TLR9 genes was compared between complicated and uncomplicated malaria (A) and between diseased young and diseased old (B). TLR4 expression was higher (P = 0.05) in complicated group (CM) in comparison with uncomplicated (UC) and no malaria (NM) groups. TLR9 expression was higher (P = 0.002) in younger age group with malaria (DY) than in the older malaria group (DO).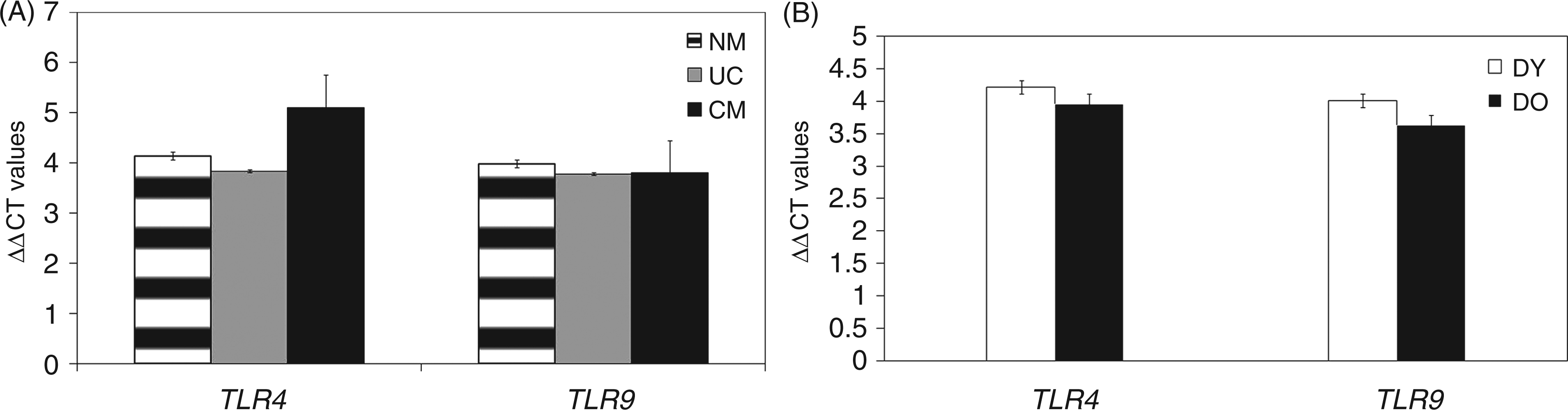
As age is an important factor in immunity to malaria, the data was analysed in the context of age. TLR9 expression was higher in the younger age group with clinical malaria (P = 0.002; Figure 2B). Expression of TLR4 gene was comparable between younger and older age groups with malaria (Figure 2B).
Discussion
Vertebrate TLRs play a crucial role in immune protection and have undergone selection in response to immune challenge, as noted in studies that have associated TLRs and their SNPs with diseases. 3 Selection favouring homozygosity or heterozygosity is one of the mechanisms that maintain polymorphism and genetic variability. Our data indicated TLR9 1486T/C heterozygosity to be protective, while it was negatively-associated with complicated malaria, whereas TLR9 (T-1237C) was seen to predispose to complicated malaria in heterozygosity. Similar findings have been reported earlier with respect to association of TLR9 (T-1237C) mutation with severe malaria. 34 However, our observations in context of (T-1486C) polymorphism are in contrast with earlier reports where it was associated with susceptibility to severe malaria in children and also with the adverse fetal outcome in women with placental malaria.15,17 Although TLR9 (T-1237C) mutation has been correlated with complicated malaria, in the presence of the (T-1486C) mutation which was associated with protection from complicated malaria, a larger proportion of uncomplicated malaria was seen in our study, suggesting that (T-1486C) was the determining mutation. Advantages conferred by heterozygosity are well documented, as in the case of sickle cell haemoglobinopathy, which affords protection against mortality from falciparum malaria in heterozygous individuals, balancing the severe consequences of the disease in homozygous individuals. 35 Similarly, it was also found that those individuals with heterozygous class I and class II HLA alleles progressed from HIV infection to an AIDS-defining illness more slowly and had the ability to clear hepatitis B infection respectively.36,37 We did not observe an association of SNPs and TLR4, either with malaria severity or frequency of clinical episodes. In contrast, Mockenhaupt et al. reported a higher frequency of TLR4 polymorphisms Asp299Gly and Thr399Ile in malaria cases, which increased the risk of severe malaria. 15 Although, the proportion of the A/A genotype of TLR4 was seen to be higher in both the ethnicities in the present study, it was, however, not associated with either protection or susceptibility to malaria. TLR4 alleles were not in HWE in the overall population, including each of the two ethnic groups. Natural selection acting on human TLR4 ascribed to the presence of excess rare non-synonymous polymorphisms has been reported by Nakajima et al., who postulated that the selection was linked to pathogen pressure. 38
Expression of TLR4 genes was enhanced in patients having complicated malaria in the present study, which could possibly be an outcome of binding to GPI anchors of parasite surface proteins leading to increased levels of pro-inflammatory cytokines and also resulting in the up-regulation of cell adhesion molecules (ICAM-1 and VCAM-1), leading to cytoadherence.5,39 An earlier study also reported increased expression of TLR4 in adults with severe and mild malaria, but decreased TLR9 expression in people with no history of malaria. 40 Also, Coban et al. found that innate immune responses via TLR2 and TLR9, and adaptor molecule MyD88-dependent pathways are critically involved in the pathogenesis of cerebral malaria. 41 On the contrary, Lepenies et al. demonstrated that induction of cerebral malaria was independent of TLR2, TLR4 and TLR9 gene expression. 42 TLR9 gene expression was seen to be age-related, with higher expression in younger malaria patients. Interestingly, the proportion of uncomplicated malaria was higher in this group and may be seen as an outcome of increased TLR9 gene expression, which induced an early pro-inflammatory response resulting in containment of the disease. In a parallel study we analysed the gene expression of a panel of cytokines (IL-2, IL-4, IL-8, IL-12α, IL-12β, IL-18, IFN-γ, TNF-α and TGF-β) and found that early pro-inflammatory cytokines were up-regulated in diseased cases in comparison with healthy controls (unpublished data). Increased TLR9 expression could be induced by HZ, a malarial pigment formed after digestion of the haemoglobin by malarial parasites. 43 It is well documented that the younger age group, generally, have higher parasitaemia and suffer more severe consequences of malaria.44,45 Increased infection would result in an increased concentration of HZ in the reticulo-endothelial system, which has been demonstrated to activate the immune system by binding to TLR9 and activating the downstream signaling pathway via MyD88, thereby resulting in increased TLR9 expression as one of the outcomes. 6 Parroche et al. also reported that HZ activates the immune system similar to Coban et al., although the mechanism of activation is different.6,43 Notably, higher parasitaemia was seen in the younger age group of our study population. However, no difference in mRNA levels was seen between control and disease, which was surprising as we had observed association between the SNPs in promoter region of TLR9 with disease. This was, however, not a unique finding, as, earlier, Fuse et al. had also found correlation of TLR9 SNPs with Japanese ulcerative colitis, but no correlation between TLR9 mRNA expression and disease. 46 Modulation of TLR9 gene expression through cis-regulatory variants offers an explanation for the lack of correlation between SNPs in the promoter region and levels of mRNA expression. 47
Genetic variations in TLRs have been linked to susceptibility to infectious diseases and these variations could be the result of selective pressure on the TLR genes in those populations living in malaria-endemic areas. Ethnicity was seen to be an important factor in the association of frequency of SNPs of TLR9 with malaria pathogenesis in the present study. The promoter region of TLR9 was seen to vary between the AAs and TBs, as confirmed by sequencing. This was not surprising as AAs (plains) in our study group belonged to tea tribes of Mundari language affinity and are genetically distinct from TBs, based on mitochondrial DNA and NRY haplotypes.23,24 Our data suggested that AA (plains) had the favourable genotype that protected them from complicated malaria as: (i) TLR9 1237T/T was seen to confer protection from complicated malaria and from frequent episodes of malaria; (ii) there was negligible TLR9 1486C/C genotype in AA, which was implicated in complicated malaria in TB. We propose that their favourable genotype could be a reflection of their longer exposure to malaria as compared with the TBs in our study group, as the AAs are believed to be the earliest inhabitants of the Indian plains. 23 We also compared the genotype frequencies of TLR9 between the two different AA groups and found that they did not differ, emphasizing the role of ethnicity. Thus, it may be postulated that the TLR9 gene may have been under selective pressure exerted by malaria. Our observation that TLR9 (T-1237C) alleles were not in HWE in TBs perhaps reflects an ongoing selection in this group, who are believed to have migrated from South China and Tibet approximately 5000–10,000 years ago.24,25
In conclusion, TLR9 (T-1237C) mutation was seen to be implicated in malaria pathogenesis. These two ethnic groups, namely AAs having genetic affinity with the earliest inhabitants of Indian plains and TBs whose origin leans more towards the Southeast Asians, have accumulated different SNPs in the TLR9 promoter region as indicated from our sequencing results. Our study demonstrated that AA (plains) had the favourable genotype with respect to TLR9 polymorphisms which protected them from malaria pathogenesis and this may be related to their longer exposure to malaria. While TLR9 overexpression was favourable, that of TLR4 was a risk factor for complicated malaria. These findings thus open the possibility of the application of TLR9 SNPs as markers of population at risk of severe malaria.
Footnotes
Funding
This work was supported by Department of Biotechnology, India (grant no. BT/HRD/01/002/2007) and Rajiv Gandhi National Fellowship, University Grants Commission, India (grant no. F.14-616(ST)/2008(SA-III)) to CES.
Acknowledgements
We would like to thank all the participants for their cooperation in the study. Thanks to Dr Mamta Chawla, National Institute of Cholera and Enteric Diseases, Indian Council Medical Research, Kolkata for her help in the work carried out. Special thanks to Dr Anurodh Agrawal for his help.