
Abstract
Translocation of microorganisms and endotoxin (LPS) across the gastrointestinal mucosa may exacerbate the inflammatory response and potentiate hepatic injury associated with hemorrhagic shock. Lipopolysaccharide binding protein (LBP) augments LPS signaling through TLR4. In addition, evidence suggests that TLR4-mediated injury in liver ischemia/reperfusion occurs through the IRF-3/MyD88 independent pathway. We hypothesized that administration of LBP inhibiting peptide, LBPK95A, given at the time of resuscitation would reduce liver inflammation and injury in a murine model of hemorrhagic shock by limiting LPS-induced activation of the MyD88 independent pathway. Hemorrhagic shock was induced in male, C57BL/6 mice; a mean arterial blood pressure of 35 mmHg was maintained for 2.5 h. LBPK95A peptide or equal volume Lactated Ringer’s solution was administered followed by fluid resuscitation. Mice were sacrificed at 2 and 6 h post-resuscitation. At 2 h, liver mRNA levels revealed a significant reduction in IFN-β, a cytokine produced via the MyD88 independent pathway, with LBPK95A treatment. However, mRNA levels of TNF-α, a cytokine associated with the MyD88 dependent pathway, were unaffected by treatment. The LBP inhibitory peptide did selectively reduce activation of TLR4 signaling via the IRF-3/MyD88 independent pathway. These results suggest that LBP promotes cytokine production through the MyD88 independent pathway during hemorrhagic shock.
Keywords
Introduction
According to the Centers for Disease Control, trauma is the leading cause of death in people aged 1–44 years and hemorrhagic shock is the second leading cause of acute deaths among injured patients. 1 Hemorrhagic shock can also lead to delayed deaths associated with the systemic inflammatory response syndrome (SIRS) and multiple organ failure (MOF). 2 The progression to SIRS and MOF occurs secondary to organ hypoxia followed by re-establishment of tissue oxygenation during fluid resuscitation. The initial ischemic phase leads to alterations in mitochondrial respiration and a reduction in ATP production. 3 This results in disruption of cellular homeostasis leading to cellular swelling and death. 3 The reperfusion phase is divided into two stages: an early phase occurring within 2 h post-reperfusion and a late phase beginning 6 h post-reperfusion. 4 During the early phase, cellular injury is induced by reactive oxygen species (ROS) as a result of the rapid change in redox state. 4 The late phase is characterized by neutrophil recruitment and activation causing direct cytotoxic cellular damage through ROS production, degranulation and nitric oxide release.3–5 These changes cause a dysregulation of pro- and anti-inflammatory mediators, including cytokines, leading to the generalized pro-inflammatory state known as SIRS. 5 MOF is the consequence of uncontrolled inflammation following hemorrhagic shock and resuscitation, and is a significant cause of late trauma mortality. 5 Treatments aimed at restoring a balanced inflammatory response could improve outcomes after hemorrhagic shock.
Clinical and experimental studies have demonstrated that translocation of bacteria and endotoxin after reperfusion of the gastrointestinal tract may exacerbate the development of pro-inflammatory conditions that lead to organ failure.5,6 Owing to its role in portal blood detoxification, the liver is commonly affected during hemorrhage/reperfusion.5,7,8 In addition, TLR4 is upregulated during ischemia/reperfusion injury in both parenchymal and non-parenchymal hepatic cells. Activation of TLR4 leads to the production of inflammatory mediators and tissue injury. 9 In fact, studies have shown that endotoxin neutralization results in decreased inflammatory responses and increased survival after ischemia-reperfusion injury.10,11 Therefore, it appears that treatments aimed at inhibiting TLR4 signaling could also help control the sequelae of hemorrhagic shock.
LBP is an acute phase protein primarily synthesized by hepatocytes. 12 LBP binds to the lipid A moiety of LPS and functions to augment LPS signaling 100–1000 fold through CD14 receptors. 12 In the presence of LBP, 1/1000th the amount of LPS is needed to trigger an inflammatory response. LBP–LPS binding to CD14 facilitates signaling through TLR4. 7 Intracellular signaling through TLR4 can occur via two pathways which result in differential cytokine/chemokine production. The MyD88 dependent pathway leads to IRAk-1 phosphorylation and TNF-α production through NF-κB and the MyD88 independent pathway results in IFN-β production through IRF3.13,14 It has been demonstrated that the MyD88 independent pathway may be particularly important in hepatic ischemia reperfusion injury.15,16 This selective signaling could also be significant to organ injury after hemorrhage and may be mediated by LBP.
Mice deficient in LBP have decreased hepatic injury in hemorrhagic shock and in acetaminophen toxicity.7,17 We have shown previously that administration of an LBP inhibitory peptide is protective in acetaminophen toxicity 18 but its efficacy in hemorrhagic shock is unknown. As the LBP inhibitory peptide could serve as a potential therapeutic agent for preventing liver damage, we investigated its use in an established, fixed pressure model of hemorrhagic shock-induced hepatic injury. 7 There were two goals for this investigation. The first was to determine whether the LBP inhibitory peptide would be efficacious in the treatment of hepatic injury associated with hemorrhagic shock. The second was to determine whether the inhibition of LBP with the peptide would differentially affect the function of the TLR4 pathways.
Materials and methods
Animals
Male C57BL/6 mice (12–16 wk) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). The mice were housed in a temperature-controlled room with a 12-h dark/light cycle and allowed food and water ad libitum. The University Committee on Use and Care of Animals approved all of the experiments.
Reagents
The LBP inhibitory peptide, LBPK95A, was synthesized by the University of Michigan Protein Structure Facility as described previously. 18 The peptide sequence RVQGRWKVRASFFK is identical to that reported for human LBP from amino acid 86–99 with the exception of a substitution of lysine 95 with alanine. 19 The mouse LBP sequence is highly homologous to human LBP and the peptide is active against mouse LBP in vivo.18–20 Ultrapure LPS (without contamination for TLR2 ligands) was obtained from Invivogen (San Diego, CA, USA).
Experimental protocols
The hemorrhagic shock model was performed as described previously. 7 Mice were fasted overnight (12 h) prior to surgery. The mice were anesthetized with pentobarbital (60 mg/kg, i.p.; Ovation Pharmaceutical, Deerfield, Il, USA) prior to cannulation of both femoral arteries with polyethelene 10 catheters. One catheter was connected to a transducer (Micro-med Inc., Louisville, KY, USA) for continuous blood pressure measurement and the second to a heparinized syringe. Blood was rapidly withdrawn into the syringe to reach a mean arterial blood pressure (MABP) of 35 ± 5 mmHg. This pressure was maintained for 2.5 h by re-infusion or withdrawal of blood as necessary. At the end of the hemorrhagic phase, LBP inhibitory peptide (peptide) (300 µg/mouse) diluted in Lactated Ringer’s solution (LRS) or equal volume LRS (vehicle) (100 µl) was administered i.v. For the reperfusion phase, animals were resuscitated with the original shed blood plus additional LRS (50% of the total shed blood volume) administered i.v. over 15 min. Mice were sedated using isoflurane via a nosecone (1–2% isoflurane in oxygen) during arousal after resuscitation. The catheters were removed, the vessels ligated and skin incision closed. The mice were then allowed to recover from anesthesia. A group of control animals (sham) were fasted and anesthetized for placement of the arterial catheters without subsequent hemorrhagic shock and reperfusion. The mice were given either LRS or LBP inhibiting peptide i.v. as described above and allowed to recover from anesthesia. In addition, a group of mice was fasted but did not undergo any surgical manipulations and treatments (no manipulation).
At 2 or 6 h post-resuscitation, mice were anesthetized with isoflurane (Vetone, Boise, ID, USA) for terminal blood collection and euthanized by cervical dislocation. The left lateral liver lobe was fixed in 10% neutral buffered formalin. The remainder of the liver was snap frozen in liquid nitrogen.
Plasma aminotransferase measurements
Plasma was stored at −80°C. Levels of alanine aminotransferase (ALT) were evaluated through a University of Michigan core facility using the Idexx Vet Test Analyzer (Model 8008; Westbrook, ME, USA). The Idexx Vet Test machine is a dry chemistry analyzer that uses a colorimetric optical system to quantify end-point and rate measurements of biochemical reactions.
Histology
Formalin-fixed liver tissue was embedded in paraffin and sections stained with hematoxylin and eosin. Quantitative analysis of tissue necrosis was performed using ImageJ analysis software v.1.42q (available as freeware from http://rsbweb.nih.gov/ij/). A slide of with one liver section was analyzed in a blind fashion at 8 × magnification. Six areas of interest were randomly selected. A single liver section was analyzed for each animal. Necrotic areas were defined as areas demonstrating nuclear pyknosis, increased cellular eosinophilia, loss of tissue architecture and normal cellular morphology (Figure 1). The total area of necrosis was expressed as a percent of total hepatic parenchyma based on the average of the % necrosis from the six fields evaluated per mouse.
Representative photomicrographs of liver histologic sections. (A) Liver from sham surgery animal which did not undergo hemorrhagic shock. (B) Liver 6 h post-hemorrhage/resuscitation demonstrating pathology consistent with hepatic necrosis. Hematoxylin and eosin stain (original magnification 40×).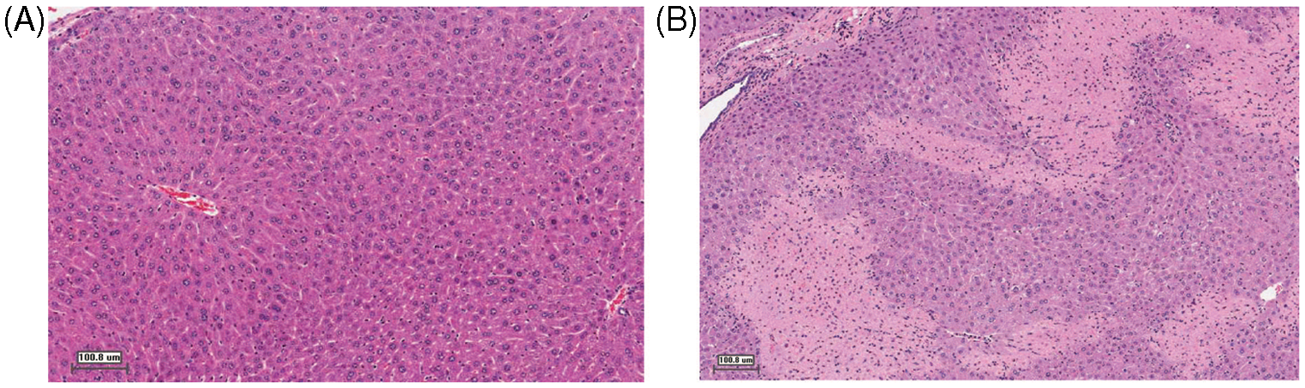
Cytokine ELISA
Cytokines were measured in plasma (1:10 dilution) and in tissue culture media (1:10 dilution) using sandwich ELISAs. Matched pairs (biotinylated and non-biotinylated) of anti-murine Abs against IL-6, TNF-α, CXCL1/KC, IP-10 and IL-10, along with their recombinant proteins (R&D Systems, Minneapolis, MN, USA) were used in methods described previously. 21 Peroxidase conjugated streptavidin (Jackson ImmunoResearch Laboratories) and the color reagent TMB (3,3′,5,5′-Tetramethylbenzidine) were used as the detection system. The reaction was stopped with 1.5 N sulfuric acid and the absorbance was read at 465 and 590 nm. 21
mRNA extraction and real-time quantitative PCR
The liver samples were stored at −80°C. Total RNA was isolated with the TRIzol® reagent as per manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). Real-time PCR was carried out as described previously. 18 Briefly, reverse transcription was performed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) to make the cDNAs. Real-time PCR was carried out using the SYBR© Green PCR Mastermix (Applied Biosystems, Carlsbad, CA, USA) and the Mastercycler© Realplex Thermal Cycler (Eppendorf, Hauppauge, NY, USA). The following primers were utilized: IFN-β: 5′-AGCTCCAAGAAAGGACGAACAT, 3′GCCCTGTAGGTGAGGTTGATCT; TNF-α: 5′GGGACAGTGACCTGGACTGT, 3′CTCCCT TTGCAGAACTCAGG IL-10: 5′GCCTTATCGGAAATGATCCA, 3′TTTTCACAGGGGAGAAATCG Actin: 5′- TCT ACG AGG GCT ATG CTC TC -3′, 5′- AAG AAG GAA GGCTGG AAA AG -3′ Melt curve analysis was performed with each PCR reaction to assure the specificity of the product.
The comparative threshold cycle (Ct) or ΔΔCt method was used to determine the relative concentration of RNA transcript between experimental samples. The results are then expressed as ‘fold changes’ relative to a calibrator. In all samples for the in vivo experiments, the calibrator used was the concentration of RNA in the wild-type control groups 2 h post-hemorrhage. For the in vitro studies, the calibrator used was the average threshold for the control untreated groups in each time point studied. The validity of the semi-quantitative method was confirmed by a consistent log linear correlation of r2 > 0.95 between starting template RNA concentration and threshold cycle for all genes studied. All values are expressed as a ratio of target amplicon to the housekeeping gene (actin).
Cell culture
1 × 106 RAW 267.4 cells (ATCC) were plated overnight in RPMI to achieve a confluency of 80–100% prior to experimentation. Cells were washed with Hank's Balanced Salt Solution (HBSS) and then cultured in RPMI + 1% wild type serum (from normal C57 BL/6 mice) with either no addition, Ultrapure LPS (Escherichia coli 0111:B4, Invivogen, San Diego, CA, USA), or LPS and LBP peptide. Cells were harvested and lysed at multiple time points after addition of LPS.
Immunoblots
Immunoblots were performed essentially as described previously, with some minor modifications. 17 Briefly, cells were washed three times with HBSS and lysed using Gold Lysis Buffer containing 20 mM Tris pH 7.9, 137 mM NaCl, 5 mM EDTA, 10% glycerol, 1% Triton X-100, 184 µg/ml sodium orthovanate and the Complete Mini protease inhibitor tablet (Roche Diagnostics, Indianapolis, IN, USA). Total protein was measured with the bicinchoninic acid (BCA) protein assay method (Pierce Chemical, Rockford, IL, USA). For immunoblots, 20 µg of total protein was loaded into each lane using of a premade 10% Precise Protein Gels (Thermo Scientific, Rockford, IL, USA), then transferred to PVDF Transfer Membrane (Thermo Scientific, Rockford IL, USA). The membranes were probed with either a primary Ab, phospho-IRF3 from Cell Signaling Technology (Danvers, MA, USA) or pIRAK-1 from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and the secondary Ab anti-rabbit HRP-linked IgG secondary (Cell Signaling Technology). Equal loading was assessed with utilizing the same blots and re-blotting after stripping and re-probing with either IRF-3 or IRAK-1 on the respective blots. In addition, all blots were also re-probed with Actin Antibody (Santa Cruz Biotechnology).
Signal detection was performed utilizing the Pierce Supersignal West Pico chemiluminescent substrate (Thermo Scientific) and quantification of the densitometry signal was performed with NIH Image J software. Relative expressions of density of the blots were calculated by dividing the area under the curve of treated by the average of the untreated groups at each time point.
Statistical analysis
Analysis was performed using Graphpad Prism software (La Jolla, CA, USA). Data are expressed as mean ± SEM. Analysis was performed using t-test or ANOVA when two or more variables were compared respectively. Statistical significance was considered to be P ≤ 0.05.
Results
MABP during hemorrhage and resuscitation
MABP was monitored before, during and after hemorrhage/resuscitation. There were no significant differences in initial MABP among the groups. Throughout the 2.5-h hypovolemic shock period, the MABP was maintained at 35.45 ± 2.028 mmHg. Post-resuscitation, MABP was comparable with initial values and was not different among groups (Figure 2). Therefore, administration of the peptide did not influence the final MABP.
Initial and final blood pressure (BP). Direct blood pressure was measured from the femoral artery of mice before induction of hemorrhagic shock (Initial) and after resuscitation (Final). Resuscitation was accompanied by administration of either peptide (300 µg/mouse) or an equal volume of vehicle (Control). There were no significant differences in blood pressure between the peptide-treated and control animals. Graph represents mean + SEM. n = 7–10/group.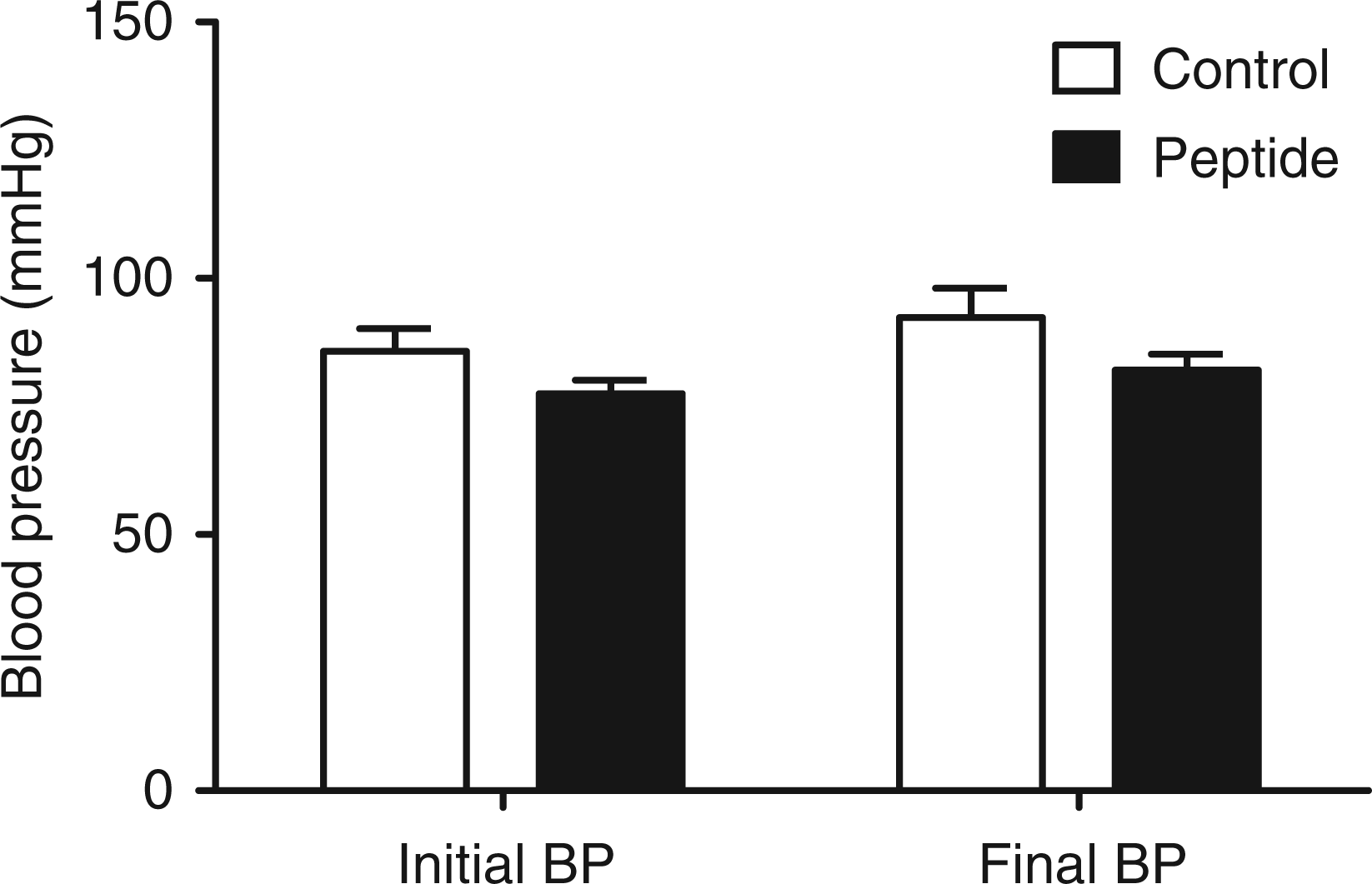
Post-hemorrhage/resuscitation liver injury
Hepatic injury increased significantly post-hemorrhagic shock with significant elevations in plasma ALT at 2 h post-hemorrhage/resuscitation compared with no manipulation animals (P = 0.0041) (Figure 3A). Plasma ALT began to decline by 6 h post-hemorrhage, but remained elevated above no manipulation and sham animal values. Similar to plasma ALT, histologic evidence of hepatic necrosis also increased by 2 h post -hemorrhage and remained increased at 6 h compared with no manipulation animals (Figure 3B).
Parameters of hepatic damage after hemorrhage/reperfusion. Groups of mice underwent no manipulations (n = 4), Sham Surgery ( + /− peptide treatment) (n = 3) or hemorrhage/reperfusion (+/− peptide) (n = 5−9). (A) Plasma ALT levels were significantly increased at 2 h compared with no manipulation animals (P = 0.0041) (B) % Hepatic necrosis was elevated at 2 and 6 h compared with sham and no manipulation animals which demonstrated no observable hepatic necrosis. Peptide treatment reduced the mean percent necrosis by 50%, although this was not statistically significant. Graph represents mean + SEM. *P < 0.05.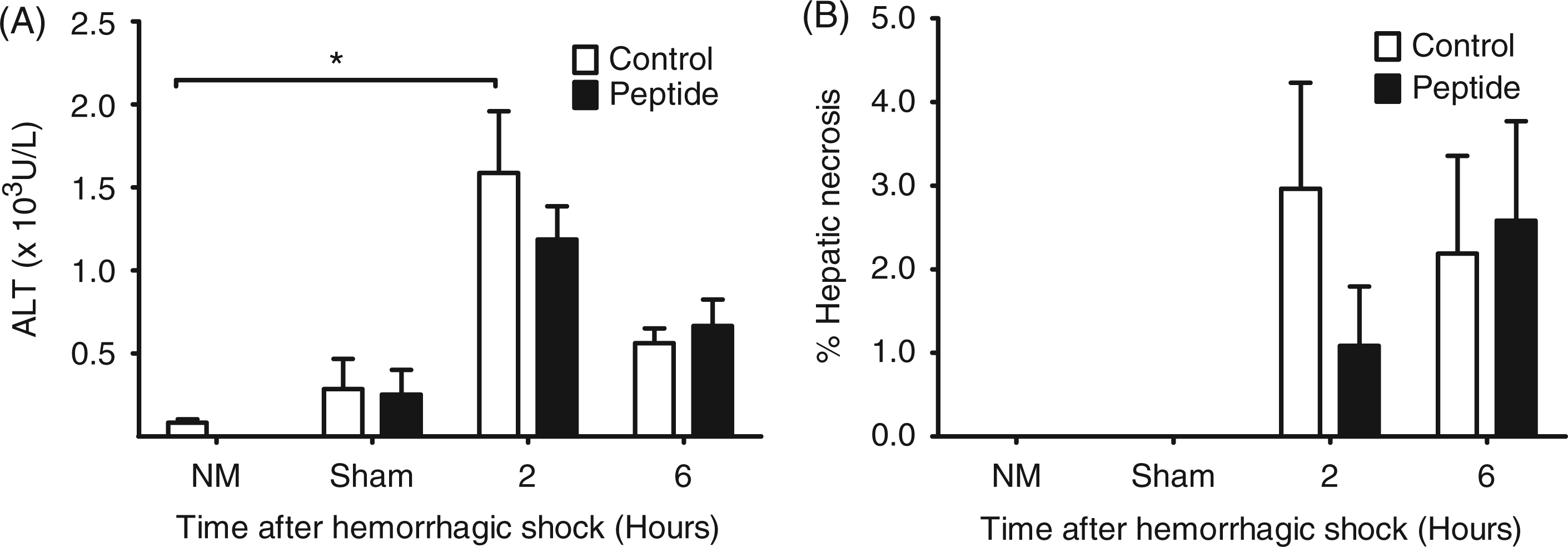
Treatment with peptide resulted in a 25% decrease in mean ALT values at the 2 h time point compared with vehicle-treated animals. Similarly, there was a ∼ 50% decrease in mean hepatic necrosis in the animals treated with the peptide compared with controls at 2 h (Figure 3B). However, these differences were not statistically significant.
Plasma cytokine levels after hemorrhage/resuscitation
In order to determine the systemic inflammatory response post-hemorrhage/reperfusion injury, we measured a panel of pro- and anti-inflammatory cytokines and chemokines (Figure 4). Plasma cytokine levels were greatest at 2 h post-hemorrhagic shock. Peptide treatment reduced the mean cytokine concentrations of the pro-inflammatory cytokines (TNF-α, IL-6 and CXCL1/KC), although the results were not significantly different from controls—likely because of variability between animals. The peptide-treated group also showed a trend toward reduction of the anti-inflammatory cytokine, IL-10.
Plasma cytokine levels after hepatic hemorrhage/reperfusion injury. Groups of mice underwent hemorrhage/reperfusion ( + /− peptide) (n = 4−9). (A) IL-6, (B) TNF-α, (C) CXCL1/KC and (D) IL-10 were highest at 2 h post-resuscitation and the mean cytokine values decreased with peptide administration. There were no statistically significant differences between treatment groups. Graphs represent mean + SEM. Parameters without error bars were below the lower limit of detection (LLD) for that cytokine.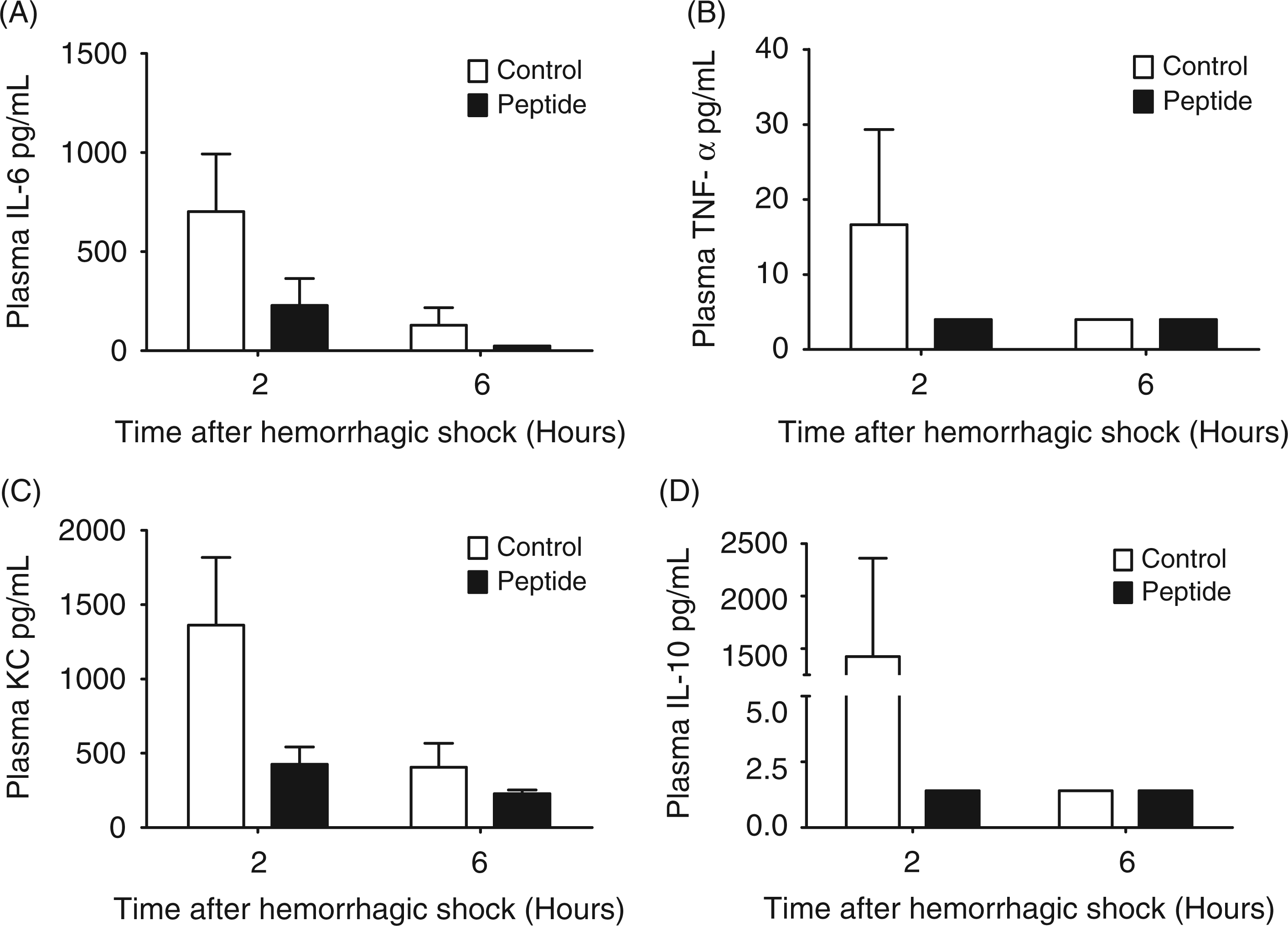
Hepatic cytokine/chemokine PCR after hemorrhage/resuscitation
In order to examine the hepatic inflammatory response to hemorrhagic shock, we examined intrahepatic RNA expression of cytokines produced by the MyD88 dependent (TNF-α) and MyD88 independent pathways (IFN-β). We found that, similar to the systemic response, cytokine expression was greatest at 2 h post-hemorrhagic shock and had declined by 6 h. Administration of peptide resulted in a significant decrease in the expression of the MyD88 independent cytokine IFN-β (P = 0.0187) but not the MyD88 dependent cytokine, TNF-α (Figure 5A). In addition to a significant decrease in the pro-inflammatory cytokine IFN-β (Figure 5B), there was also a significant decrease in intrahepatic expression of the anti-inflammatory cytokine, IL-10 (P = 0.0179) (Figure 5C).
Relative messenger RNA levels of intrahepatic cytokine expression after hepatic hemorrhage/reperfusion injury. Groups of mice underwent no manipulations (n = 4), Sham Surgery (+/− peptide treatment) (n = 3) or hemorrhage/reperfusion (+/− peptide) (n = 4−6). (A) TNF-α was significantly elevated at 2 and 6 h post-hemorrhage (control) compared with shams (control) and no manipulation animals (P < 0.05), which did not have detectable hepatic mRNA levels. There were no statistical differences between treatment groups. (B) IFN-β was significantly elevated in 2 h post-hemorrhage (control) compared with sham animals (control) (P = 0.0312 respectively) and no manipulation animals (P = 0.0133); peptide treatment resulted in a significant decline in IFN-β expression (P = 0.0.0187) at 2 h post-hemorrhage compared with controls. (C) IL-10 levels were significantly elevated in 2 h post-hemorrhage (control) compared with no manipulation animals (P = 0.0338) and treatment significantly decreased this to within normal levels (P = 0.0338) (*P < 0.05). Baseline levels of cytokines were measured 2 h post-resuscitation in vehicle control animals. Graph represents mean + SEM.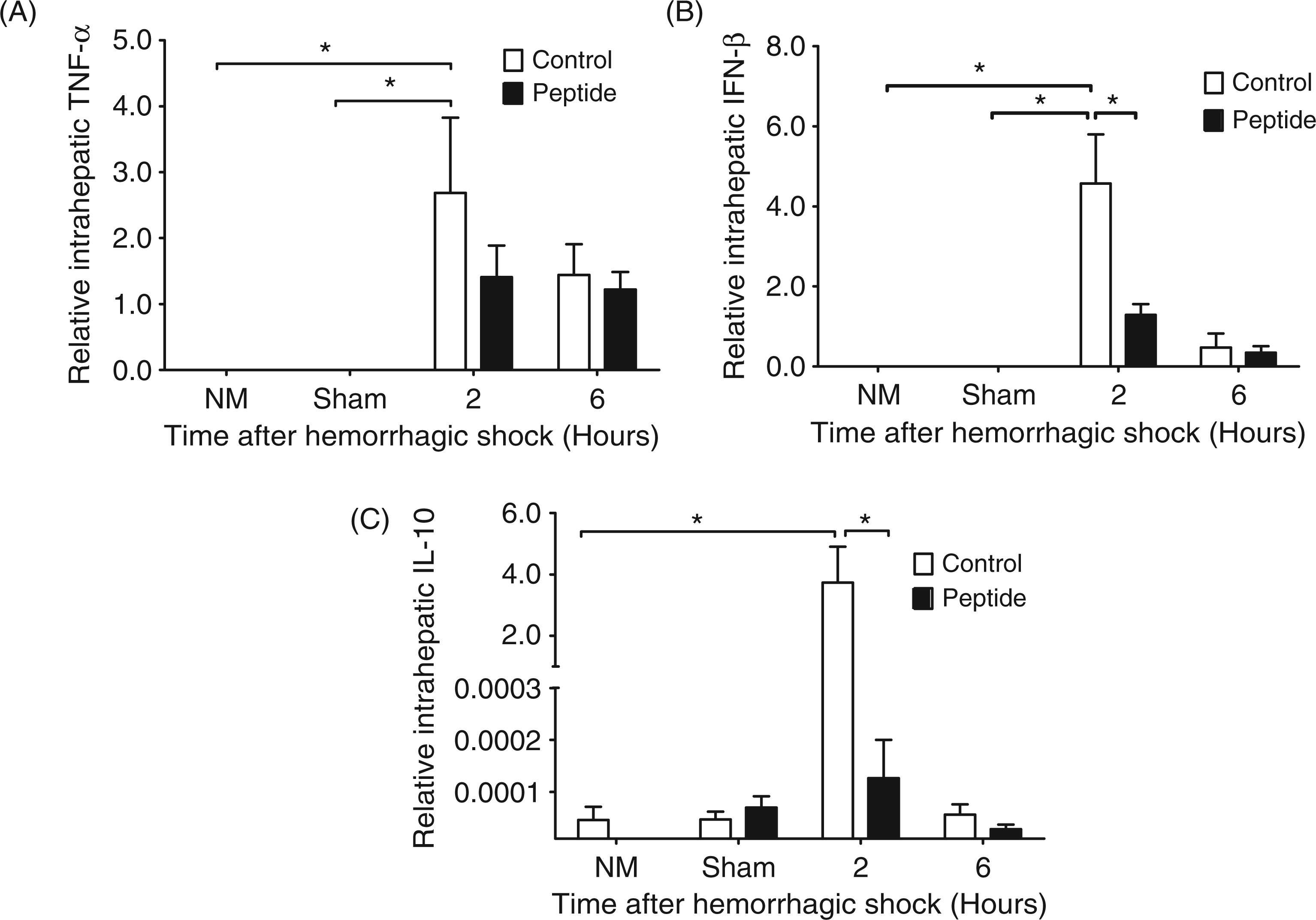
Differential effect of LBP on LPS-mediated TLR4 activation
In order to determine if the LBP inhibitory peptide differentially inhibited TLR4 activation through the MyD88 dependent and independent pathways, we examined the effect of LBP inhibitory peptide on LPS activation of IRAK-1 and IRF-3 in RAW 267.4 cells. We found that upon stimulation with LPS (10 ng/ml), there was evidence for robust phosphorylation of IRF-3 at 1 h after stimulation which peaked at 2 h (Figure 6A). Quantitative analysis of pIRF-3 levels relative to IRF-3 was significantly increased from baseline control after stimulation with LPS by 2 h (Figure 6B). Addition of LBP inhibitory peptide reduced levels of pIRF-3 in response to LPS back to baseline control unstimulated levels. In contrast, the level of phosphorylated IRAK-1 (pIRAK-1) was very low and very transient in nature (Figure 6C). The addition of LBP inhibitory peptide did not appear to inhibit the phosphorylation of IRAK-1 after LPS, although the variability between experiments made this difficult to assess (Figure 6D). Constitutive IRAK-1 levels decreased significantly after LPS, suggestive of IRAK-1 degradation (Figure 6C and Figure 6E). This degradation was inhibited by the addition of LBP peptide.
Differential effect of LBP peptide on LPS activation of MyD88 dependent and independent pathways in RAW 264.7 cells. RAW 264.7 cells cultured in 1% wild type (C57BL/6) serum and stimulated with either control media (CTRL), 10 ng/ml of LPS (LPS) or LPS with 80 µg/ml of LBP inhibitory peptide (PEP). (A) Representative immunoblots of phosphorylated IRF-3 (pIRF-3), IRF-3 (IRF-3) or actin (Actin) 1 and 2 h after stimulation. (B) Densitometry of pIRF-3/IRF-3 at 1 and 2 h after stimulation (*P < 0.05 compared with CTRL, n = 3 experiments) (C) Representative immunoblots of phosphorylated IRAK-1 (pIRAK-1), IRAK-1 (IRAK-1), and actin at 1 and 2 h after stimulation. (D) Densitometry of pIRAK-1/Actin (n = 3 experiments) and (E) densitometry of IRAK-1/Actin (*P < 0.05 compared with CTRL, n = 3 experiments).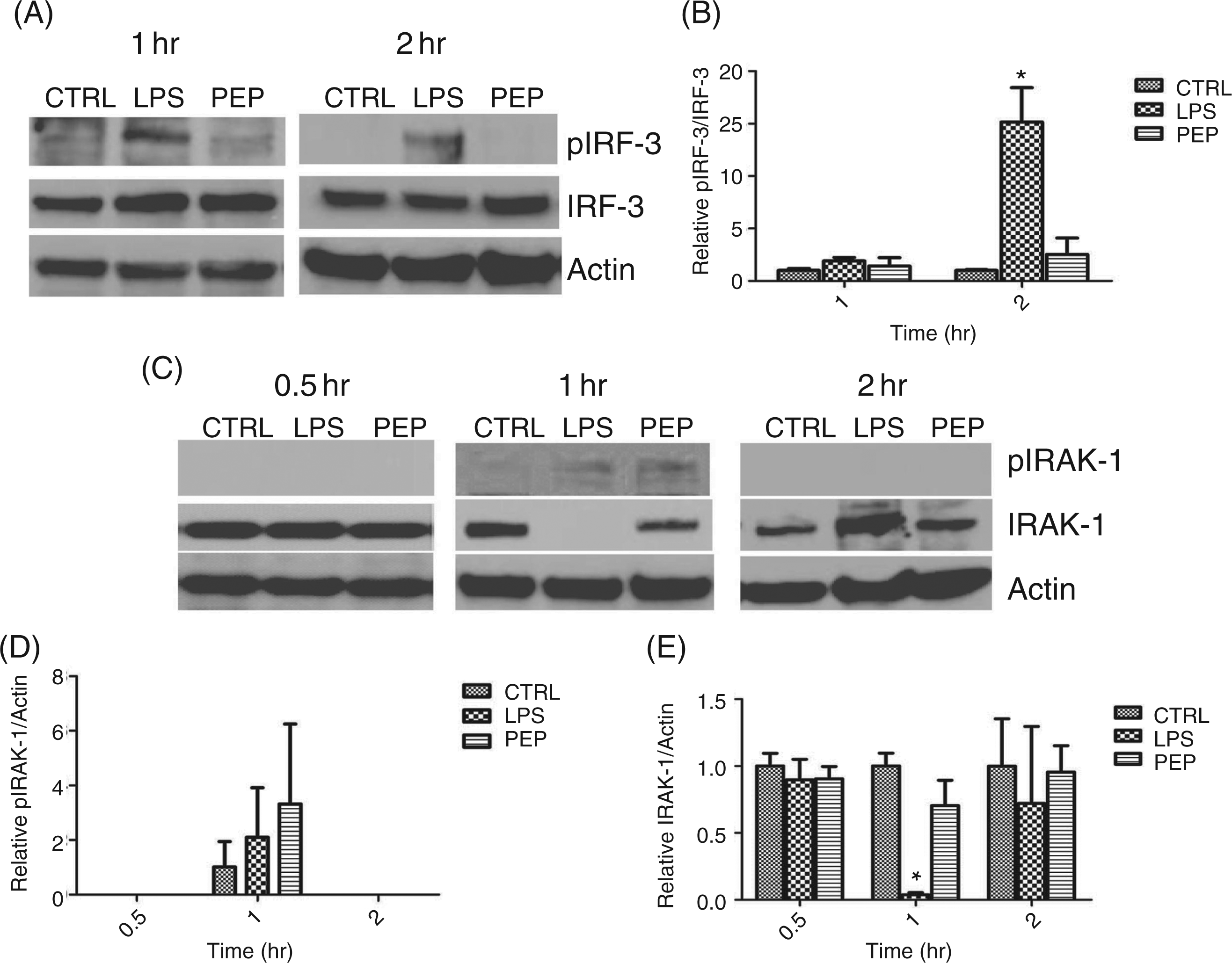
In examining differential cytokine expression by RAW 267.4 cells after LPS, we found that the levels of TNF-α RNA increased significantly by 2 h 4–5-fold compared with baseline untreated (CTRL) levels (Figure 7A). The addition of LBP inhibitory peptide inhibited this rise in TNF-α expression at 2 h such that levels of TNF-α were back to baseline, but not at 6 h. Similarly, RAW 264.7 IFN-β RNA levels significantly increased after LPS. However, the level of elevation was up to 140-fold higher than baseline CTRL levels and peaked at 2 h after stimulation but persisted at 6 h after stimulation (Figure 7B). The addition of LBP inhibited LPS-stimulated IFN-β RNA levels at 2 h and 6 h. Cumulative protein levels of TNF-α and IP-10 (as a surrogate marker for MyD88 independent stimulation by LPS) measured by ELISA also showed significant elevations at 6 h after stimulation which was inhibited by the LBP inhibitory peptide (Figure 7C and 7D).
Cytokine expression by RAW 267.4 cells after stimulation with LPS. RAW 264.7 cells cultured in 1% wild type (C57BL/6) serum and stimulated with either control media (CTRL), 10 ng/ml of LPS (LPS) or LPS with 80 µg/ml of LBP peptide (PEP). (A) Relative expression of TNF-α by RAW 264.7 cells was measured with real-time PCR at 0.5, 1,2, or 6 h after stimulation (*P < 0.05 compared with CTRL untreated cells, n = 3 experiments per group) (B) Relative expression of IFN-β by RAW 264.7 cells was measured with real-time PCR at 0.5, 1, 2, or 6 h after stimulation (*P < 0.05 compared with CTRL untreated cells, n = 3 experiments per group) (C) Cumulative expression of TNF-α was measure with ELISA at 6 h after stimulation (*P < 0.05 compared with CTRL untreated cells, n = 3 experiments per group) (D) Cumulative expression of IP-10 was measured with ELISA at 6 h after stimulation (*P < 0.05 compared with CTRL untreated cells, n = 3 experiments per group).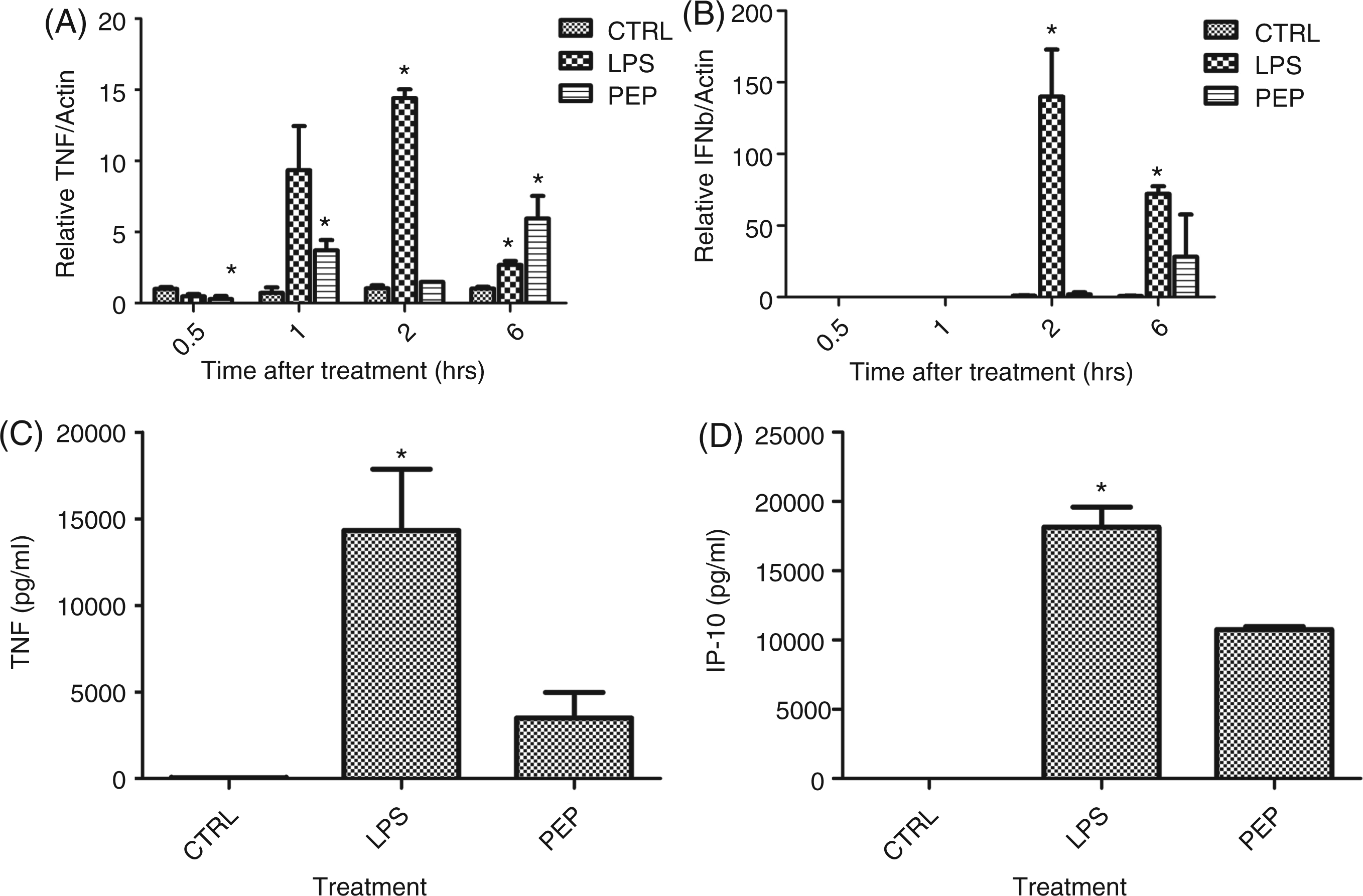
Discussion
In this study we investigated the inflammatory response in post-hemorrhage/reperfusion mice receiving LBP inhibitory peptide, LBPK95A, at resuscitation. Previous studies have demonstrated bacterial translocation and portal endotoxemia after hemorrhage/resuscitation in a similar model to the one used in this study.7,11 The released endotoxin signals through TLR4, which results in production of inflammatory mediators. 22 As the portal for gut endotoxin and site of the largest population of resident macrophages, the liver is a major source for both pro- and anti-inflammatory cytokines.23,24 In the present study, the treatment with LBP inhibitor peptide resulted in an overall trend towards reduced systemic, pro-inflammatory cytokine levels (IL-6, CXCL1/KC and TNF-α). Reducing systemic inflammation may translate to a positive effect on SIRS. In addition, there was a decrease in systemic IL-10 production. The anti-inflammatory effects of IL-10 are well known and a reduction in IL-10 could potentially counteract the anti-inflammatory effects of the LBP inhibitory peptide. However, IL-10 has also been implicated as a major mediator of the immunosuppression seen after hemorrhagic shock. IL-10 promotes the less protective Th2 polarization of T cells which may increase susceptibility to infection. 25 Therefore, the LBP inhibitory peptide may promote a more balanced cytokine response by reducing excessive production of both pro- and anti-inflammatory cytokines.25,26
In addition to the systemic inflammatory response, local hepatic inflammation was also evaluated after hemorrhage/reperfusion. Peptide treatment resulted in significant changes in the pattern of intrahepatic cytokine expression 2 h post-resuscitation. Peptide treated animals displayed significantly lower IFN-β hepatic mRNA levels than were observed in the control animals. Previous experiments have demonstrated that LBP plays a critical role in IFN-β expression via TLR4 signaling and in vivo production of MyD88 independent end-products only occurred in the presence of LPS and serum containing LBP. 27 In contrast, in our study, the prototypical MyD88 dependent TNF-α expression was not effectively blocked by LBP inhibition. This suggests that signaling through TLR4 was differentially modulated by the inhibition of LBP. Further, LBP inhibitory peptide significantly lowered expression of the anti-inflammatory cytokine, IL-10. Studies have shown that LPS-induced IL-10 expression requires the presence of IFN-β. 28 Therefore, this supports our observation of a significant decrease in IL-10 concurrent with a reduction in IFN-β when LPS signaling was inhibited. Also, IL-10 can negatively regulate TLR4 MyD88-dependent signaling through transcriptional, post-transcriptional and post-translational regulation of cytokine production, and subsequently reduction in pro-inflammatory mediators through this pathway.29,30 The lower IL-10 expression seen after peptide treatment may result in more TNF-α expression. This is an alternative explanation for why TNF-α expression was not greatly affected by LBP inhibition. Overall, peptide administration resulted in significant modulation of hepatic inflammatory mediators leading to decreased production in both pro- and anti-inflammatory cytokines. Concurrent with the peak hepatic inflammatory response, there was an increase in liver injury after hemorrhage and reperfusion. Activation through the MyD88 independent pathway of TLR4 signaling has been shown to be important in the development of hepatic necrosis secondary to ischemia/reperfusion injury. IRF-3 deficient mice have attenuated hepatic injury when compared with controls. In contrast, MyD88-deficient mice experienced the same degree of hepatic necrosis as controls. 15 However, in this study, LBP inhibitory peptide administration prior to resuscitation did not confer hepatoprotection. This is in conflict with studies of hemorrhagic shock in LBP-deficient mice in which the lack of LBP activity resulted in a reduction in hepatic injury. 7 There are several possible explanations for this discrepancy between studies.
Zhai et al. 16 demonstrated comparable findings when administering recombinant bacterial/permeability-increasing (BPI), a protein functionally related to LBP, in a model of warm ischemia/reperfusion. Animals treated with BPI did not have a difference in serum ALT or hepatocellular damage compared with controls, 16 suggesting that endogenous TLR4 ligands other than endotoxin may be involved in the early inflammatory signaling observed during ischemia reperfusion injury. Endogenous ligands coupled with endotoxin may be synergistically responsible for hepatic ischemia/ reperfusion injury and blocking endotoxin alone may not be sufficient to reduce hepatic injury. 16 In addition, peptide administration occurred at the time of reperfusion and the timing of administration may be important to hepatoprotection. Although the timing of treatment with the LBP inhibitory peptide was clinically relevant, it did not allow blockade of LBP during the hypotensive period of the injury. Studies have demonstrated that the release of endotoxin after hemorrhage is early after resuscitation and transient, with a 50% decrease in levels within 30 min of resuscitation. 7 Therefore, the principal hepatic insult may have already occurred by the time treatment was instituted.
In order to completely examine the effects of LBP peptide on LPS-stimulated effects through the MyD88 dependent and independent pathways, we examined its effects on the RAW 264.7 macrophage-like cell line. LPS signaling through the TLR4 pathway results in MyD88 recruitment and interaction with IRAK-1 resulting in phosphorylation of IRAK-1 and activation of the TRAF-6 leading to the downstream activation of NF-κβ and TNF-α production. Simultaneous activation also occurs via the TRIF-TRAM pathway, leading to IRF-3 phosphorylation and IFN-β production. Because of the redundancy in signal with the TRIF-TRAM pathways and interactions of TRAF 6 leading to late NF-κβ activation, we chose to look at signaling upstream to TRAF-6 to differentiate the effects on the two pathways. We found that IRAK-1 phosphorylation was very transient in response to LPS and was not clearly inhibited by the peptide. Furthermore, constitutive IRAK-1 levels diminished with LPS stimulation consistent with prior reports that LPS induced IRAK-1 phosphorylation results in rapid degradation of IRAK-1 through the ubiquitin/protesome pathway. 31 This is consistent with the brisk increase in TNF-α expression that was inhibited by the LBP peptide. In contrast to its effect on the MyD88 dependent pathway, the effect of LBP on the MyD88 independent pathway was more robust and prolonged. We found that IRF-3 phosphorylation was significantly diminished with the addition of the peptide and the effect of the IFN-β signal was more robust and prolonged. The studies in the RAW 267.4 cells illustrate the differential effects of LBP on LPS stimulation via the two pathways. Similar to the in vivo data, the LBP inhibitory peptide had a prolonged and significant effect on IFN-β, while the effect on the TNF-α RNA levels was much more brisk and transient. Because both the MyD88 dependent and independent pathways can affect TNF-α RNA levels, and because there are many different cellular sources of TNF-α other than macrophages, the combined effects seen in vivo show a trend towards decrease of TNF-α with LBP inhibitory peptide and a profound and significant reduction in IFN-β.
Treatment of hemorrhagic shock with the LBP inhibitory peptide, LBPK95A, at the time of resuscitation did selectively reduce activation of TLR4 signaling via the IRF-3/MyD88 independent pathway, suggesting LBP promotes cytokine production through the MyD88 independent pathway during hemorrhagic shock. Although not completely hepatoprotective in this model of hemorrhagic shock, targeted inhibition of LPS signaling may prove useful for combined control of pro- and anti-inflammatory responses to hemorrhagic shock and reperfusion.
Footnotes
Funding
This work was supported in part by a Veterans Administration Merit Award (G.L.S).