
Abstract
TLRs play a key role in innate immune defenses. It was previously reported that purified respiratory syncytial virus (RSV) fusion protein elicits an inflammatory response in hematopoietic cells, which required expression of TLR4 and its co-receptor CD14. However, a biological role of TLR4 in immunity to RSV, as initially proposed, has remained inconclusive and controversial. Here, we directly assess the role of human TLR4 and its co-receptors in NF-κB activation, viral entry and replication using intact virions rather than purified RSV components. We used HEK 293 reporter cells that are highly permissive for RSV and that either express or a lack a functional human TLR4/MD-2/CD14 complex. We demonstrate that RSV-mediated NF-κB activation, viral entry and replication are independent of the expression of a functional human TLR4/MD-2/CD14 complex and that, in turn, human TLR4 activation by LPS remains unaffected in RSV-infected cells. Thus, although isolated viral compounds such as purified RSV F protein may bind TLR4 and/or CD14, a direct interaction between intact RSV particles and the human TLR4 receptor complex does not seem to play a biological role in RSV pathogenesis.
Keywords
Introduction
Human respiratory syncytial virus (RSV) is the leading cause of acute lower respiratory infection in children under 5 years of age and is also an important respiratory pathogen in the elderly, asthmatics and immunocompromised hosts, causing considerable morbidity and mortality among these risk groups.1–4 An RSV vaccine is not currently available. In the 1960s, clinical trials that aimed to assess the classic approach of immunizing children using formalin-inactivated RSV (FI-RSV) were abandoned as it became clear during the following winter (RSV season) that FI-RSV-vaccinated children were not protected. The hospitalization rate upon RSV infection actually increased to 80% (16 out of 20 children) among the FI-RSV-vaccinated children in comparison to 5% (1 out of 21 children) of the control group that had received a parainfluenza vaccine, and two FI-RSV-vaccinated children died.5,6
Severe RSV disease is likely a consequence of excessive airway inflammation, involving the expression and release of several cytokines and chemokines that are regulated by NF-κB.7–11 What drives NF-κB-dependent cytokine secretion in response to RSV is incompletely understood. NF-κB can be activated upon stimulation of a variety of receptors, including pattern recognition receptors, such as TLRs and retinoic acid-inducible gene (RIG)-like receptors, as well as cytokine receptors, such as the TNF receptor (TNFR) and the IL-1 receptor (IL-1R).12,13
Kurt-Jones et al. 14 reported that purified RSV Fusion (F) protein elicits a marked (and the G and N proteins a weak) increase in secretion of several pro-inflammatory cytokines in hematopoietic cells and that RSV F protein-induced IL-6 secretion in murine peritoneal macrophages is dependent upon functional TLR4 and CD14. Thus, TLR4-mediated signaling has been implicated in innate immune responses and the outcome of RSV infection. However, Lizundia et al. 15 did not find a role for purified RSV F protein in stimulating TLR4-dependent activation of NF-κB. Similarly, contradictory results concerning the role of TLR4 in RSV infection come from several studies in the mouse model. Whereas some of these studies14,16,17 argue that RSV elicits TLR4-dependent immune responses in mice, others did not find any clinical, virological or immunologic parameters of RSV infection altered in the absence of TLR4 18 and suggested that (inactivated) RSV is a poor TLR agonist. 19 Indeed, Delgado et al. 19 attributed the failure of the FI-RSV vaccine trials during the 1960s to poor TLR stimulation by inactivated RSV, leading to the generation of pathologic Abs with low affinity and consequently enhanced disease upon subsequent infection. Addition of purified TLR4 agonists to UV- or FI-RSV vaccines is protective against enhanced disease in different animal models,19–21 further suggesting that entire (inactivated) RSV does not activate TLR4. Of note, the initial results in the mouse model arguing for a role of TLR4 activation during RSV pathogenesis14,16 were confounded by an additional loss-of-function mutation in the IL-12 R β-chain in the TLR4-deficient mouse strains,18,22 as well as by the use of cell surface markers with insufficient cell type specificity. 18
The impact of common non-synonymous single nucleotide polymorphisms (SNPs) in the human TLR4 gene on disease outcomes in human subjects infected with RSV also remains controversial. Tal et al. 23 and Awomoyi et al. 24 reported an over-representation of a heterozygous genotype in two commonly co-segregating TLR4 polymorphisms (TLR4 Asp299Gly and Thr399Ile) among term infants with severe RSV infection and among primarily premature infants with symptomatic RSV infection, respectively. However, neither of these two association studies was confirmed in an independent cohort. In contrast, in our previous studies 25 we did not find an association between the same TLR4 polymorphisms and the risk of severe RSV infection using an independent cohort of children and comparing subjects with severe RSV infection (in patients) to subjects with mild RSV infection or a general population control. 25 We also demonstrated in vitro that these TLR4 polymorphisms have no impact on LPS or RSV-induced cytokine responses in PBMCs from a pediatric population, regardless of whether or not the children were stratified by asthmatic status or prior bronchiolitis.25,26
Study of isolated viral components (purified RSV F protein) as undertaken by Kurt-Jones et al. 14 does not allow the assessment of the specific biological role of TLR4 in RSV pathogenesis, nor can it substitute the study of intact virions. These observations can be interpreted as a role of TLR4 either in RSV-mediated NF-κB activation or in viral entry, which is largely mediated by the action of the RSV F protein. 27
Results from mouse infection models and use of primary cells from TLR4-deficient mice to assess the role of TLR4 in RSV pathogenesis have been confounded by the presence of second site mutations. 18 Importantly, human and murine TLR4 do not have identical ligand specificities.28–30 Therefore, the goal of this study was to assess directly the role of human TLR4 in NF-κB activation and viral entry by using cells of identical origin that either express or lack a functional human TLR4/MD-2/CD14 complex. In our studies, we used stably transfected HEK 293 cells. In contrast to hematopoietic cells, HEK 293 cells do not express functional TLRs endogenously and are highly permissive for RSV. We also used intact virions rather than isolated RSV components. We demonstrate that expression of a functional human TLR4/MD-2/CD14 complex has no impact in RSV-mediated NF-κB activation, viral entry and replication and that, in turn, RSV infection has no effect on human TLR4 activation by canonical TLR4 agonists.
Materials and methods
Cell culture and reagents
HEK 293 reporter cell lines, namely TLR4-positive cells (HEK-Blue™ hTLR4, InvivoGen, San Diego, CA, USA) and TLR4-negative cells (HEK-Blue™ Null2, InvivoGen, San Diego, CA, USA), as well as 1HAEo− cells (kindly provided by Darryl Knight, UBC James Hogg Research Centre, Providence Heart and Lung Institute at St. Paul's Hospital, Vancouver, BC, Canada) were grown in DMEM, High Glucose (HyClone, Logan, UT, USA) with 2 m
Virus preparation
RSV A2 and Long were obtained from the American Type Culture Collection. Strain rgRSV30 31 was generously provided by Mark Peeples (Nationwide Children’s Hospital, Columbus, OH, USA). RSV serotype B strain 9320 and RSV serotype A community isolate HLI 1 32 were generously provided by David Marchant (UBC James Hogg Research Centre, Providence Heart and Lung Institute at St. Paul's Hospital). All RSV strains were propagated in HEp2 cells (Hybrid Diagnostics) for 48–72 h and virus particles were precipitated from the cell culture supernatant by use of polyethylene glycol as described elsewhere. 33 We avoided sonicating or freeze-thawing the RSV-infected cells to minimize contamination by cellular components. For some experiments the virus was further purified by ultracentrifugation on a discontinuous sucrose gradient. 33 All virus preparations were aliquoted, quick frozen and stored in liquid nitrogen until used. Virus titers were determined on HEp2 cells by end-point dilution and counting syncytia stained indirectly either using a mouse anti-RSV F protein monoclonal Ab (Novus Biologicals), a secondary HRP-conjugated anti-mouse IgG Ab (Cell Signaling, Danvers, MA, USA) and Liquid DAB+ Substrate Chromogen System (Dako), or by use of a FITC-conjugated mouse anti-RSV F protein monoclonal Ab (Chemicon International).
Purified TLR ligands
Ultrapure LPS from Escherichia coli K-12 and serotype O111:B4 (TLR4 agonists), and LPS from Rhodobacter sphaeroides (TLR4 antagonist), were from InvivoGen. LPS from Bordetella pertussis strain 18-323 (TLR4 antagonist) was prepared as described previously. 28
HEK 293 reporter cell assays
Fifty thousand cells were seeded in each well of a tissue-culture treated, 96-well, flat-bottom, polystyrene plate in the presence of selective antibiotics. The next day, cells were gently washed and then incubated in fresh media (without selective antibiotics) either containing RSV, purified LPS or without stimulus as indicated. For each experiment, all conditions were done in triplicate. Secreted embryonic alkaline phosphatase (SEAP) activity was measured in the culture supernatant with QuantiBlue substrate (InvivoGen) according to the manufacturer’s instructions.
Determination of infectivity via flow cytometry
Two hundred thousand cells were seeded in each well of a tissue-culture treated, 24-well, flat-bottom, polystyrene plate and infected the next day with 50,000 plaque forming units (pfu) of rgRSV30 per well (titrated on HEp2 cells) in the presence or absence of 10 ng/ml ultrapure LPS from E. coli K-12. 24 and 48 h past infection (hpi), cells were detached with Hanks'-based, enzyme-free cell dissociation buffer (Gibco), washed once with D-PBS containing 0.5% BSA, and re-suspended in the same buffer. For each experiment, all conditions were done in duplicate. Virus-infected, GFP-expressing cells were quantified with a FACSCalibur flow cytometer (BD Biosciences).
Statistical analysis
Replicates within each single experiment were averaged and then analyzed across multiple experiments using statistical software (GraphPad Prism). The number of independently performed experiments is stated in each Figure. Statistical comparisons of NF-κB-dependent SEAP activity between TLR4-positive and TLR4-negative cells were performed using two-way ANOVA with repeated measures and Bonferroni post-tests. Comparisons of RSV infectivity in HEK 293, 1HAEo− and THP1 cells to that in HEp2 cells were done using one-way ANOVA and Bonferroni post-tests. P < 0.05 was considered significant and indicated wherever significant differences were observed.
Results
Live RSV fails to activate NF-κB in a TLR4-dependent manner
To assess the role of the human TLR4/MD-2/CD14 complex in RSV-mediated NF-κB activation, we utilized a HEK 293 cell line that was stably transfected with plasmids encoding an NF-κB-inducible SEAP and a constitutively expressed human TLR4/MD-2/CD14 complex (hTLR4-positive cells). To control for TLR4-independent responses, we included the stably transfected parental HEK 293 cell line in our assays that only harbors the NF-κB-inducible SEAP reporter but lacks a functional TLR4/MD-2/CD14 complex (hTLR4-negative cells). Both cell lines were either infected with the commonly used RSV strain A2 at different multiplicities of infection (MOI) ranging from 0.002 to 2 or stimulated with different doses of purified LPS from E. coli K-12, and reporter activity was measured 24 hpi. Upon infection with RSV A2, low NF-κB-dependent reporter activity was only detectable at a high infection dose (MOI of 2) but this response was not dependent upon expression of a functional TLR4/MD-2/CD14 complex as we detected a similar response in both hTLR4-positive and hTLR4-negative cells (Figure 1A). In contrast, stimulation of the hTLR4-positive cells with purified LPS from E. coli K-12 showed NF-κB-inducible reporter activity in a dose-dependent manner that was significantly different from the TLR4-negative cells at doses as low as 1 ng/ml (Figure 1A). Similarly, despite the responsiveness of the TLR4-positive cells to very low doses of LPS, we found no significant differences in NF-κB-inducible reporter activity between TLR4-positive and TLR4-negative cells 24 and 48 hpi after stimulation with a highly purified preparation of RSV strain A2 (by means of sucrose density gradient ultracentrifugation) and when using several other RSV strains, including strain Long, RSV serotype B strain 9320, and community isolate HLI1
32
(Figure 1B, C). As RSV strain A2 has been used by Kurt-Jones et al.,
14
Haynes et al.
16
and Haeberle et al.
17
to support a role of TLR4 in RSV pathogenesis, we chose to use A2 wild type or A2-derived recombinant strains in all further studies.
Expression of a functional TLR4/MD-2/CD14 complex does not influence NF-κB activation in response to RSV. HEK 293 reporter cells that either expressed (hTLR4-positive) or lacked expression of a functional TLR4/MD-2/CD14 complex (hTLR4-negative) were exposed to different doses of purified LPS from E. coli K-12 or preparations of live RSV. (A) RSV strain A2 isolated from culture fluids of infected HEp-2 cells by precipitation with polyethylene glycol. (B) and (C) RSV serotype A strains Long, HLI1 and serotype B strain 9320 that were similarly isolated, and RSV A2 that was further purified by ultracentrifugation on a discontinuous sucrose density gradient (RSV A2purif.). NF-κB-induced SEAP reporter activity was assessed after 24 h (A, B) and 48 h (C) using QUANTI-Blue™ substrate and by reading the OD655. Shown are mean differences and SEM in the OD655 readings relative to unstimulated cells. Data are from three or more independent experiments, each done in triplicate. *P < 0.05, **P < 0.01, ***P < 0.001.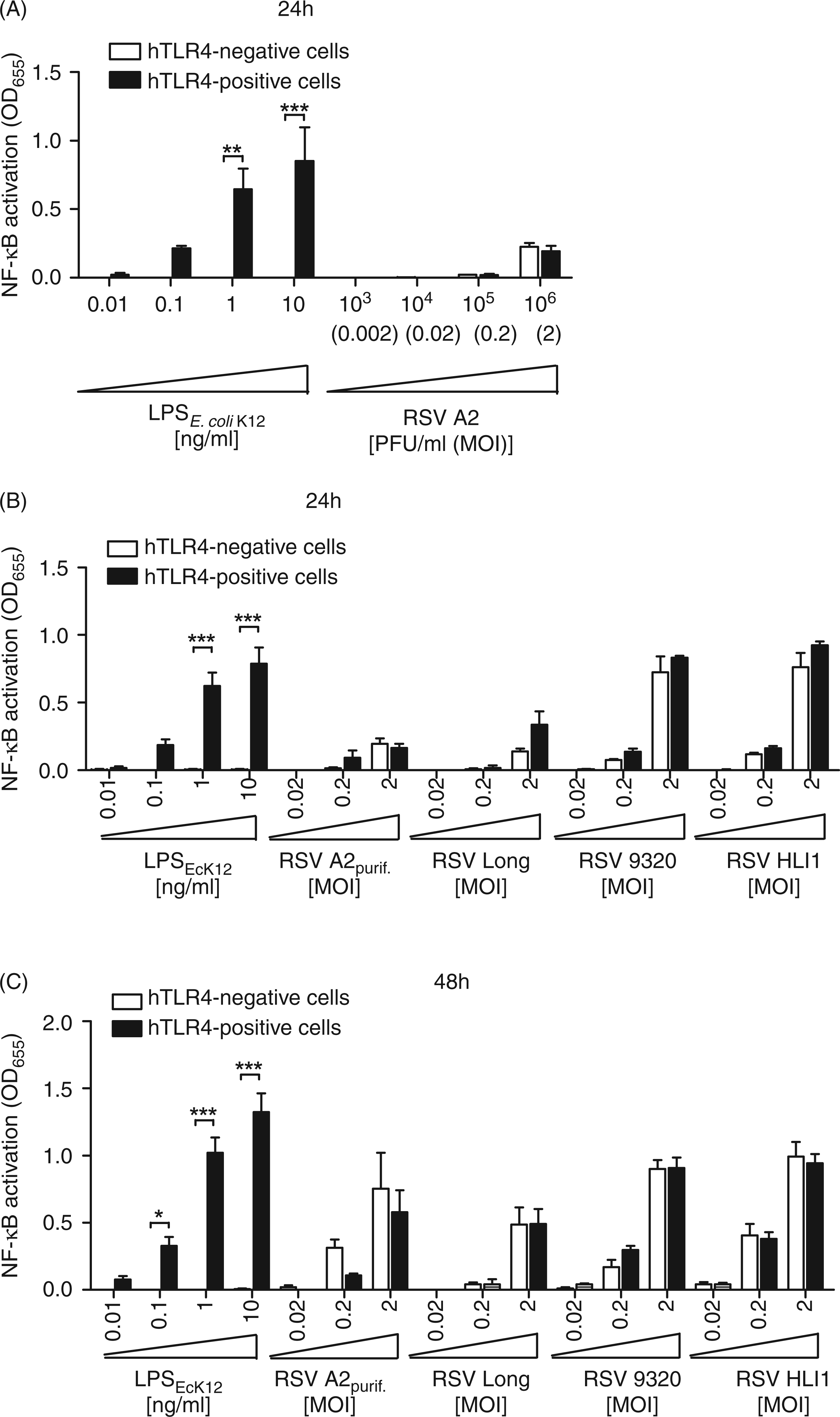
The TLR4/MD-2/CD14 complex plays no role in RSV entry and replication
Next, we examined the role of TLR4 in RSV entry and replication. We reasoned that if interaction of the RSV F protein with TLR4 plays a biological role in viral attachment and entry, then cells expressing a functional TLR4/MD-2/CD14 complex would be significantly more permissive for RSV than those that lack functional TLR4. For this purpose, we infected TLR4-positive and TLR4-negative HEK 293 cells with similar doses of sucrose density gradient-purified rgRSV30 in the presence or absence of 10 ng/ml ultrapure LPS from E. coli K-12 to induce TLR4-mediated signaling. Strain rgRSV30 is a recombinant RSV A2 derivative that expresses Renilla GFP under control of a viral promoter.
31
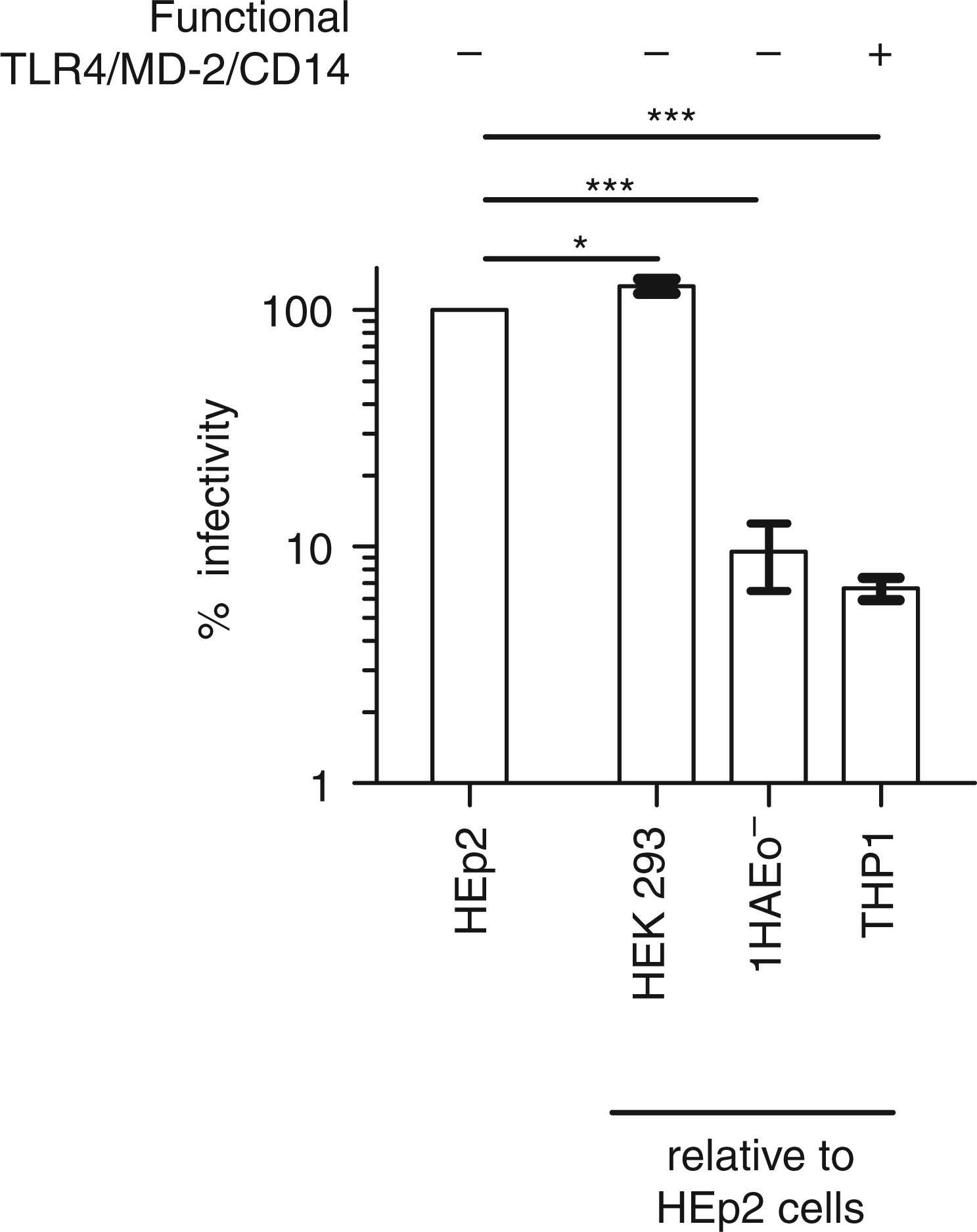
Fluorescence microscopy and flow cytometry was used to detect infected, GFP-expressing cells 24 and 48 hpi. We found that the number of rgRSV30-infected cells 24 and 48 hpi was not significantly influenced by the presence of a functional TLR4/MD-2/CD14 complex, and that stimulation of TLR4-mediated signaling with LPS from E. coli K-12 also did not alter the infectivity (Figure 2). However, we observed a significant increase of rgRSV30-infected cells from 24 to 48 hpi and a visible cytopathic effect at 48 hpi in both cell lines (Figure 2), confirming that RSV infects and replicates in HEK 293 cells. We also compared rgRSV30 infectivity in (TLR4-negative) HEK 293 cells, 1HAEo− cells (a bronchial epithelial cell line) and THP1-derived macrophages with the infectivity in HEp2 cells (commonly used to propagate and titrate RSV), because these cells also differ in their ability to express a functional TLR4/MD-2/CD14 complex. As shown in Figure 3, HEK 293 cells are slightly more permissive for RSV than HEp2 cells, but infectivity in 1HAEo− cells and THP1-derived macrophages is significantly (>10-fold) lower. Importantly, only in THP1-derived macrophages, but not in HEp2 or in 1HAEo− cells, can secretion of pro-inflammatory cytokines (IL-6 and IL-8) be induced upon exposure to high doses (1 µg/ml) of purified LPS from E. coli K-12. These data provide empiric evidence that HEp2 and 1HAEo− cells lack a functional TLR4/MD-2/CD14 complex and further demonstrate that RSV infectivity is not dependent upon a functional TLR4/MD-2/CD14 complex.
Expression of a functional TLR4/MD-2/CD14 complex does not influence RSV infectivity. HEK 293 reporter cells that either expressed (hTLR4-positive) or lacked expression of a functional TLR4/MD-2/CD14 complex (hTLR4-negative) were infected with rgRSV30 (recombinant RSV A2-derived strain that expresses GFP) in the presence or absence of 10 ng/ml purified LPS from E. coli K-12. Cytopathic effects were assessed 24 and 48 hpi by fluorescence microscopy (top) and rgRSV30-infected (GFP stained) cells were quantified using flow cytometry (bottom). The micrographs (top) show bright field images merged with images taken through filters for GFP detection. The bar diagram (bottom) depicts means and SEM of % GFP-stained cells. Data are from four (hTLR4-negative and hTLR4-positive cells without LPS) or three (hTLR4-positive cells + LPS from E. coli K-12) independent experiments, each done in duplicate. Infectivity of rgRSV30 in HEK 293 cells (hTLR4- negative), 1HAEo− cells and THP1-derived macrophages was compared with that in HEp2 cells by end-point dilution and counting GFP-stained cell clusters (syncytia) 48 hpi. Shown are mean % values ± SEM of normalized data (HEp2 = 100%). Data are from four (HEp2 and HEK 293 cells) or three (1HAEo− and THP1 cells) independent experiments, each done in triplicate. *P < 0.5, ***P < 0.001.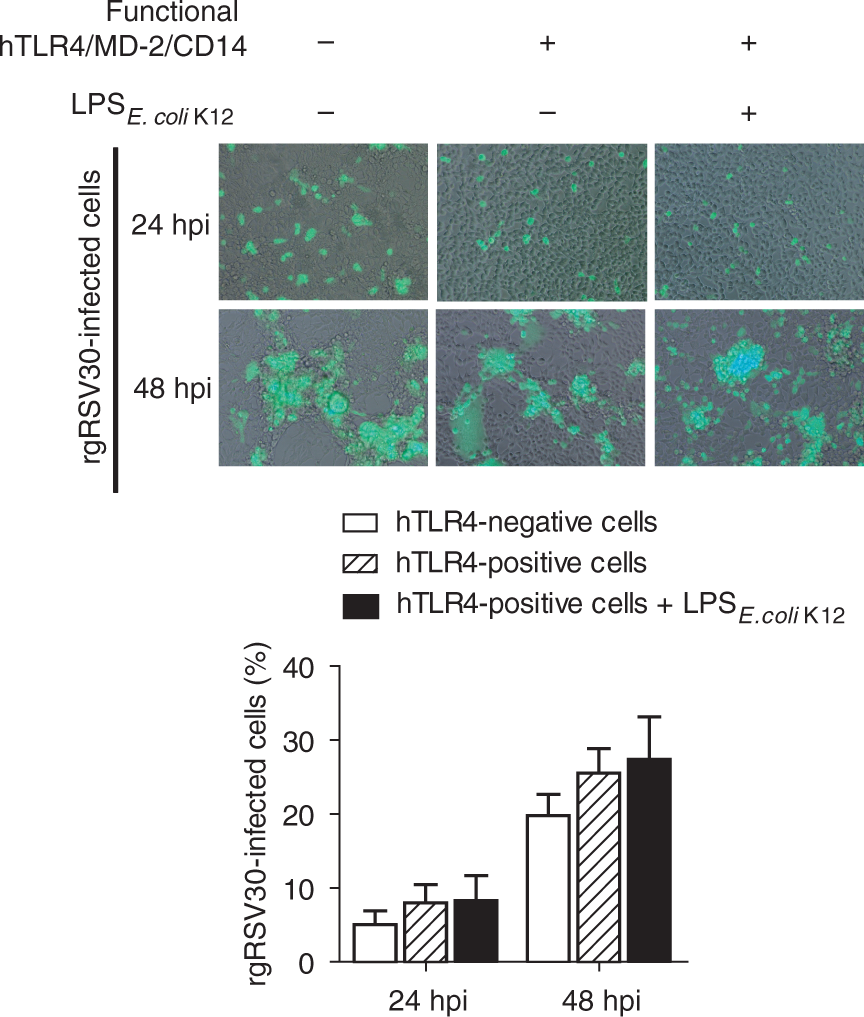
RSV infection does not interfere with ligand-specific, CD14-independent and CD14-dependent TLR4 activation
Finally, we asked whether or not RSV infection interferes with LPS-specific activation of the TLR4/MD-2/CD14 complex. TLR4 activation by LPS from E. coli proceeds by insertion of 5 of the 6 lipid A acyl chains in a deep hydrophobic pocket of MD2, causing local conformational changes in MD-2 which triggers hydrophilic interactions of MD2 with TLR4, as well as interactions of the lipid A phosphate groups and the remaining exposed acyl chain with TLR4, thereby inducing the formation of symmetrical TLR4/MD-2 heterodimers and phosphorylation of the intracellular signaling domains of TLR4.
34
We reasoned that if intact virions interact directly with either TLR4 or its co-receptor molecule CD14, biological activity of canonical TLR4 agonists such as E. coli LPS would be compromised in RSV-infected cells. For this purpose, we stimulated TLR4-positive HEK 293 cells with a constant low dose of LPS from E. coli K-12 (1 ng/ml) sufficient to induce NF-κB-dependent SEAP reporter activity in these cells (see Figure 1), and simultaneously infected the cells with increasing doses of RSV A2 or added increasing doses of known human TLR4 antagonists (purified LPS from R. sphaeroides
35
and B. pertussis strain 18-323).
28
As shown in Figure 4A, we found that in contrast to the human TLR4 antagonists, RSV is unable to abolish the biological activity of LPS from E. coli K-12, a canonical TLR4 agonist. As Kurt-Jones et al.
14
reported that purified RSV F protein-mediated cytokine induction also requires the expression of CD14 and because LPS of E. coli K-12 lacks a repetitive O-polysaccharide and therefore does not require CD14 for TLR4-mediated signal induction,
36
we repeated these experiments using 10 ng/ml LPS isolated from E. coli O111:B4 instead of LPS of E. coli K-12. LPS of E. coli O111:B4 has a repetitive O-polysaccharide and therefore requires the accessory protein CD14 for TLR4-mediated NF-κB activation.
36
Again, we found that in contrast to known human TLR4 antagonists, RSV is unable to abolish the biological activity of LPS from E. coli O111:B4 (Figure 4B).
RSV infection does not influence TLR4 activation by its canonical agonists. HEK 293 reporter cells that express a functional TLR4/MD-2/CD14 complex (hTLR4-positive) were stimulated with 1 ng/ml purified LPS from E. coli K-12 (A) or 10 ng/ml purified LPS from E. coli O111:B4 (B), alone or in combination with increasing doses of either RSV A2 or purified hTLR4 antagonists (antagonist 1 = LPS from R. sphaeroides; antagonist 2 = LPS from B. pertussis 18-323). NF-κB-induced SEAP activity was assessed 24 hpi using QUANTI-Blue™ and by reading the OD655. Shown are mean OD655 differences and SEM in the OD655 readings relative to cell-free medium. Data are from three (A) or two (B) independent experiments, each done in triplicate.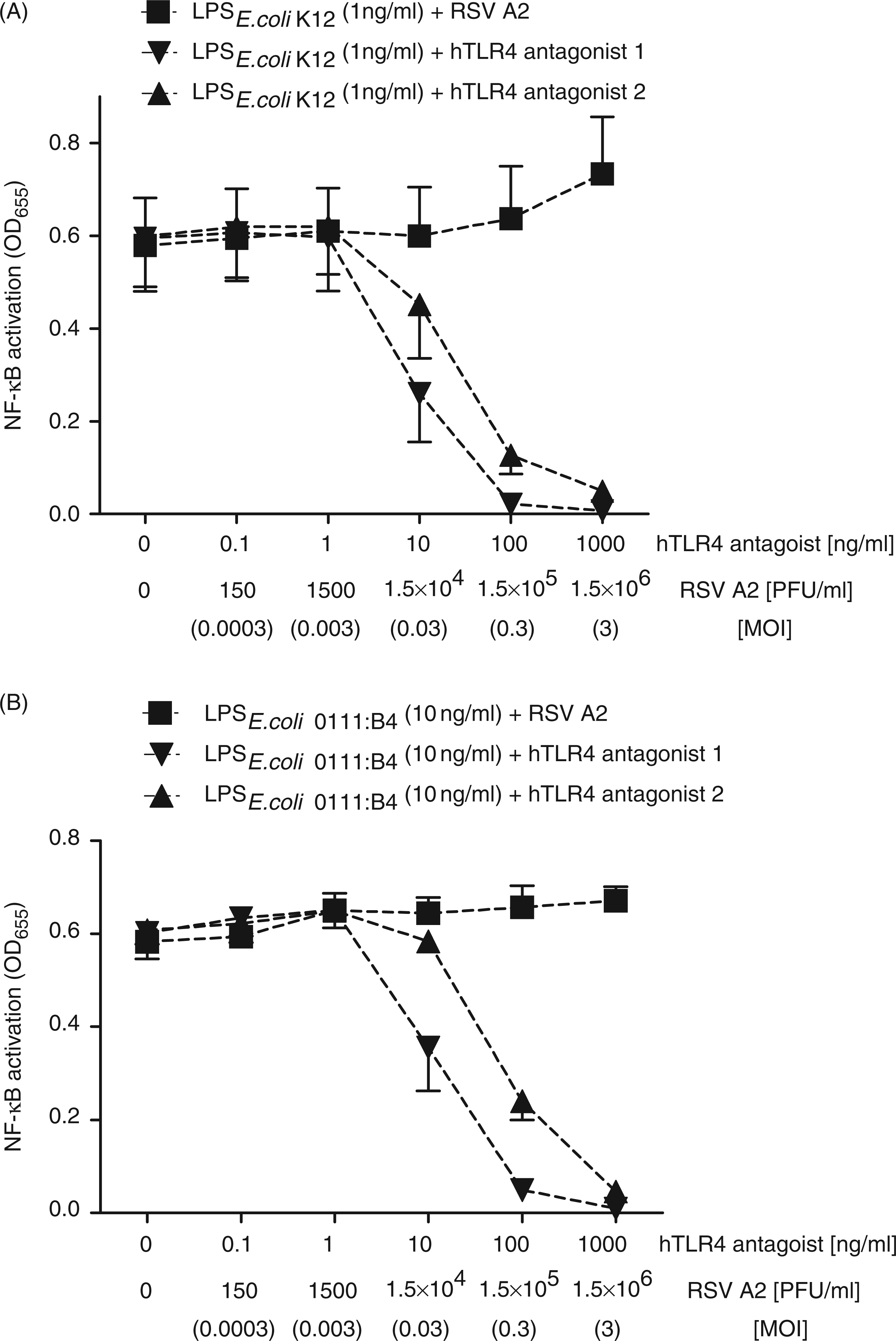
Discussion
Our results demonstrate that expression of a functional human TLR4/MD-2/CD14 complex does not play a role in NF-κB activation mediated by infectious RSV particles. This is inconsistent with the role of TLR4 in innate immune responses to RSV, as suggested by Kurt-Jones et al. 14 who reported that RSV F protein-induced IL-6 secretion in mouse peritoneal macrophages depends upon functional TLR4 and CD14. Of note, Kurt-Jones et al. 14 used RSV A2 propagated in Vero (African green monkey kidney) cells instead of the commonly used HEp2 (human laryngeal carcinoma) cells to isolate the RSV F protein. This technical difference may be relevant as it has recently been shown that propagation of RSV in Vero cells results in expression of truncated attachment (G) protein and 600-fold lower infectivity of the virus in human airway epithelial cells. 37 The RSV F protein is not naturally released by the virions or RSV-infected cells but remains membrane-associated. 38 Kurt-Jones et al. 14 isolated RSV F protein by lysis of RSV-infected cells in a non-denaturing detergent buffer followed by chromatography over an affinity column containing RSV F protein-specific Ab. Although the authors 14 have conclusively shown that RSV F protein-induced pro-inflammatory cytokine secretion was not caused by contaminating bacterial LPS, they did not address a potential role of contamination by endogenous TLR4 ligands—so-called damage-associated molecular patterns (DAMPs),39,40 or other inflammatory mediators that could have potentially confounded their results. The latter could explain why Kurt-Jones et al. 14 also observed cytokine production by monocytic cells in response to similarly isolated RSV G and N proteins, albeit weaker than with the F protein. To minimize effects caused by contaminating cellular components, we isolated the virions from culture supernatants of RSV-infected HEp2 cells while avoiding sonication or freeze-thaw cycles, and also used sucrose-purified RSV preparations in our study.
More difficult to explain and reconcile are two other studies which reported that RSV-infected TLR4 knock-out mice exhibited reduced NF-κB activity in lung tissue 17 and failed to express alternative peritoneal macrophage markers 41 in comparison with RSV-infected wild-type mice. There are a number of variables through which TLR4 may indirectly influence the outcome of RSV infection. For example, TLR4 activation can alter normal lung development42,43 and risk of atopy is influenced by genetic variation in the trimolecular complex of TLR4/MD-2/CD14. 44 Thus, differences in immune signaling between RSV-infected wild-type and TLR4-deficient mice may not be caused by direct interaction of RSV with the TLR4-receptor complex, but rather different cytokine profiles or differences in lung development induced by LPS from the environment or by the natural bacterial gut flora, and that these differences are being amplified during RSV infection. Further studies are needed to fully elucidate the role of TLR4 in RSV pathogenesis.
Nevertheless, our data are consistent with the findings of Delgado et al. 19 who demonstrated that the lack of Ab affinity maturation in mice that were inoculated with UV-inactivated RSV was a consequence of poor TLR stimulation, which, in turn, led to enhanced RSV disease upon subsequent challenge with live RSV. Indeed, the authors explained why the FI-RSV vaccine used in clinical trials during the 1960s did not protect the children but instead let to severe disease and they demonstrated that supplementation of UV-inactivated RSV with LPS or other TLR ligands let to DC maturation, T helper activation and B cell affinity maturation, thereby protecting against enhanced disease upon subsequent challenge. 19 Similar observations were made by others, demonstrating that the addition of the TLR4 agonist monophosphoryl lipid A to a FI-RSV vaccine prevented excessive cytokine release and reduced or eliminated leukocyte infiltration within the alveoli of cotton rats post-challenge with RSV.20,21 These findings argue against a role of the RSV F protein in TLR4 activation and suggest that enhanced RSV disease is rather a consequence of a failed TLR activation. Our data are also consistent with that of Ehl et al. 18 who did not find any clinical, virologic or immunologic parameters of RSV infection in mice altered in the absence of TLR4. Nevertheless, it cannot be ruled out that TLRs other than TLR4 become activated upon exposure to live RSV. Delgado et al. 19 have shown that both the Ab avidity for the RSV F protein and the RSV-neutralizing capacity of Abs elicited by i.n. inoculation of mice with live RSV were decreased in MyD88+/− mice in comparison with wild-type mice. Others have suggested a role of TLR7/8 in RSV-induced delay in neutrophil apoptosis. 45
We also demonstrate that RSV infectivity and expression of a functional TLR4/MD-2/CD14 complex are not correlated, indicating that TLR4 function is not required for RSV entry. Marchant et al. 46 had initially suggested a role of TLR4 in RSV entry because they observed in 1HAEo−cells that p38 MAPK is activated during virus entry and that TLR4 clusters at the site of virus-host cell contact. Therefore, the authors concluded that TLR4-mediated activation of p38 MAPK is a determinant of RSV entry. However, Marchant et al. 46 did not assess whether or not the TLR4 protein that they detected by fluorescence microscopy in 1HAEo− cells is functional, which we found not to be the case using the same cells. Marchant et al. 46 were also not able to conclusively demonstrate that RSV entry can be blocked by using TLR4-specific neutralizing Abs. In a later study 32 the same group demonstrated that cell-surface nucleolin, a ubiquitous protein expressed by virtually all cell types, acts as the RSV receptor. Nucleolin directly interacts with the RSV F protein, and RSV entry and replication can be abolished by siRNA-mediated knock-down or polyclonal Abs directed against cell-surface nucleolin. 32
Finally, we demonstrate that RSV infection has no impact on LPS-induced TLR4 activation, regardless of whether or not co-receptor CD14 is required. Thus, although isolated viral compounds may directly bind to TLR4 and/or CD14 thereby inducing pro-inflammatory cytokines, this does not seem the case for intact virions. Interestingly, Shingai et al. 47 have found that a soluble, naturally released form of the RSV G protein 38 inhibits TRIF-dependent IFN-β induction via an as yet unknown mechanism acting downstream of TLR4 and 3 but not downstream of RIG-I or MDA5. 47 In our study, we investigated NF-κB activation which primarily regulates pro-inflammatory cytokine secretion in non-hematopoietic cells and we used RSV preparations that contained enriched virions while soluble proteins, culture media and host cell components were removed during virus isolation. This likely explains why we observed no interference of RSV particles with ligand-specific, CD14-independent and CD14-dependent TLR4 activation. Therefore, we conclude there is no biologically relevant protein–protein interaction between intact RSV particles and the human TLR4 receptor complex.26,29,30
Footnotes
Funding
This work was supported by funding from the Canadian Institutes of Health Research Team in Mutagenesis and Infectious Diseases. N.M. is a recipient of AllerGen's Canadian Allergy and Immune Diseases Advanced Training Initiative award and a fellowship co-funded by the Canadian Institutes of Health Research, the Canadian Lung Association and GlaxoSmithKline Inc. S.E.T. is a clinical scholar of the Michael Smith Foundation for Health Research.
Acknowledgements
We thank Mark Peeples and Peter Collins for generously providing recombinant RSV strain rgRSV30 and David Marchant for RSV strains 9320 and HLI1. We thank Martine Caroff and Alexey Novikov for generously providing highly purified LPS of B. pertussis strain 18-323.
Conflicts of interest
The authors have no conflicts of interest to declare.