
Abstract
Innate immunity is crucial for an effective host defense against pathogenic microorganisms in periodontal tissues. As periodontal ligament (PDL) cells synthesize immunomodulatory cytokines, the aim of this in vitro study was to investigate whether these cells can interact with innate immune cells. Resting and inflammatory primed (IL-1β, TNF-α, HMGB1) human PDL cells were co-cultured with human monocyte-derived dendritic cells or macrophages. Migration, phenotypic maturation and modulation of phagocytosis of Porphyromonas gingivalis by immune cells were investigated upon co-culture with PDL cells and/or their released soluble factors. PDL cells interacted with immune cells under both non-inflammatory and inflammatory conditions. Immune cell migration was significantly enhanced by co-culture with PDL cells, which also affected their phenotypic maturation both through cell-cell contact and through released soluble mediators. The dendritic cell maturation markers CD83 and CD86 were upregulated as much as both ‘alternatively activated’ M2 macrophage maturation markers CD23 and CD163. In contrast, the ‘classically activated’ M1 macrophage maturation marker CD64 was downregulated. Finally, PDL cells significantly enhanced the phagocytosis of Porphyromonas gingivalis by immune cells. Our experiments revealed that PDL cells are not only structural elements of the periodontium, but actively influence immune responses by interaction with innate immune cells.
Introduction
Cells of the innate immune system are crucial for an effective host defense against pathogenic microorganisms in periodontal tissues. However, distorted immune responses caused by over-activated immunoinflammatory mechanisms lead to hard and soft periodontal tissue destruction by a chronic local immune response. 1 Pathophysiologic mechanisms involve cellular components and soluble products, such as antimicrobial peptides, complement fragments, cytokines and chemokines, that lead to the development of cell-mediated and humoral immune responses.
Periodontal disease is characterized by chronic inflammation that is initiated by accumulated plaque and the presence of anaerobic bacteria in the periodontal pocket, which results in an inflammatory reaction and the progressive loss of periodontal ligament and alveolar bone.2,3
Periodontitis progression involves the expression of pro-inflammatory cytokines, such as IL-1β and TNF-α. 4 Additionally, recent studies found out that HMGB1, which is already known to play an important role in inflammation, is expressed much more pronounced in periodontally diseased tissues and the gingival crevicular fluid from patients with chronic periodontitis compared to periodontally healthy subjects.5,6
Chemokines associated with periodontitis pathogenesis are predominantly synthesized by resident cells of the periodontal tissues, which thereby sustain and enhance the migration of immune cells to sites of inflammation.1,4,7 Consequently, migration of these professional antigen-presenting cells (APCs) into oral tissues as a first line of defense against pathogenic microorganisms is essential for the initiation of an adequate immune response. They perform their sentinel function by recognition of danger-associated signals through pattern recognition receptors that sense pathogen-derived molecules and subsequently undergo functional maturation.8–11
Dendritic cells (DCs) are professional APCs of the innate immune system with potent Ag-capture and Ag-presenting functions, but are also able to induce tolerance or Ag-specific unresponsiveness, which is dependent on the cytokine microenvironment of the periphery.12–15 In their immature stage, immature DCs (iDCs) reside as sentinel cells in the epithelium and the interstitium of most solid organs. Characteristics of mature DCs (mDCs) are the loss of phagocytic capacity, as well as the upregulation of co-stimulatory molecules and MHC class II. 16
Macrophages are another entity of cells involved in innate immune responses with distinct physiological properties depending on the tissue they are located in and on the cytokines that induce their maturation. 17 Resident macrophages develop from circulating myeloid precursors under the influence of cytokines and chemokines during their recruitment into tissues. 18 Specific inflammatory mediators released upon tissue damage or infection can phenotypically polarize these resident macrophages into ‘classically activated’ M1 or ‘alternatively activated’ M2 macrophages (below defined as ‘M1’ and ‘M2’), which is accompanied by the formation of distinct immunologic properties.19–20 M1 polarization is primarily affected by IFN-γ and pathogen-associated molecular patterns, whereas IL-4 and IL-13 trigger an M2 phenotype. 21 However, macrophages are thought to possess functional plasticity that implies the potential to reversibly adapt to changes in their microenvironment. This involves the development of tissue- and response-specific functional patterns with pro- or anti-inflammatory, immunogenic or tolerogenic, and tissue-destructive versus tissue-restorative activities.22,23
Raising evidence suggests that fibroblasts, which are known to produce paracrine immune modulators, are crucially involved in inflammation control and in the regulation of immune responses.24,25 Periodontal ligament (PDL) cells represent the main cellular constituent of the PDL and are known to synthesize immunomodulatory cytokines that are supposed to influence the local response to infections.26,27
It was the aim of this study to investigate whether PDL cells interact with cells of the innate immune system by influencing migration and differentiation of DCs and macrophages both through cell–cell interactions and the release of soluble factors. Furthermore, the phagocytic capacities of DCs and macrophages were examined in a simulated inflammatory periodontal environment with regard to an effective host defense against periodontal pathogens.
Material and methods
The study was conducted in full accordance with ethical principles, including the World Medical Association Declaration of Helsinki; the experiments were undertaken with the understanding and written consent of each subject. The study was independently reviewed and approved by the ethics committee of the University of Bonn.
Cytokines, Abs and pathogens
Cytokines included recombinant human IL-1β (5 ng/ml; PromoCell, Heidelberg, Germany), recombinant human TNF-α (5 ng/ml; Biozol Diagnostica, Eching, Germany) and recombinant human high-mobility-group-protein B1 (HMGB1; 100 ng/ml; GenWay Biotech Inc., San Diego, CA, USA).
FACS Abs were purchased from BioLegend (San Diego, CA, USA) and included APC anti-human CD11c, FITC anti-human CD1a, phycoerythrin (PE) anti-human CD83, Pacific Blue™ anti-human CD86, Pacific Blue anti-human CD14, FITC anti-human CD23, PE anti-human CD163 and APC anti-human CD64. Carboxyfluorescein diacetate succinimidyl ester (CFSE) was obtained from GenWay Biotech Inc.
Porphyromonas (P.) gingivalis 381 was heat inactivated at 95°C for 10 min and harvested from liquid culture by centrifugation. Bacterial pellets were washed threefold in sterile PBS. Optical density (OD)600 was determined using a Biochrom WPA CO8000 cell density meter (Biochrom Ltd., Cambridge, UK), at which a value of 0.1 equaled approximately 108 P. gingivalis cells per ml. 28 Then, the bacteria were conjugated to pH-sensitive pHrodo™ rhodamine-based fluorogenic dyes as a specific sensor of phagocytosis with the pHrodo™ Phagocytosis Particle Labeling Kit for flow cytometry (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
PDL cell culture
Cultures of human PDL cells from six periodontally healthy donors were explanted from the middle third of the root surface of teeth removed during routine extraction for orthodontic reasons. The teeth were extracted from adolescent patients after examination of defined variables for clinically healthy periodontal tissues with the absence of bleeding on probing, probing depth <4 mm and loss of attachment level <3 mm.
PDL cells were grown in DMEM (Invitrogen) supplemented with 10% heat inactivated FCS (Invitrogen) and 1 µg/ml penicillin/streptomycin (Invitrogen) at 37 °C in a humidified 5% CO2 atmosphere. After reaching confluence, cells of the fourth passage were passaged using trypsin/EDTA (Invitrogen) and used for experiments in the fifth passage.
DC and macrophage culture
Human monocytes were purified from buffy coats of healthy human donors (provided by the blood bank of the University of Bonn) in compliance with institutional review board protocols using Ficoll/Paque (Life Technologies Inc., Carlsbad, CA, USA) density gradient centrifugation of heparinized blood. Monocytes were generated in vitro by seeding PBMCs in X-VIVO20 medium (Cambrex Bio Science, Walkersville Inc., Walkersville, MD, USA).
After 2 h of incubation at 37 °C, non-adherent cells were removed and the adherent blood monocytes were further cultured in DMEM supplemented with 10% heat inactivated FCS and 1 µg/ml penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere.
Generation of monocyte-derived iDCs was obtained by adjunct of the cytokines recombinant human GM-CSF (100 ng/ml; Berlex Laboratories, Bothell, WA, USA) and recombinant human IL-4 (20 ng/ml; R&D, Minneapolis, MN, USA) every other day for 7 d in culture.
Generation of monocyte-derived macrophages (below defined as ‘M0’) was obtained by adjunct of the cytokine recombinant human GM-CSF every other day for 7 d in culture. The quality and purity of cell preparation was routinely controlled by morphologic analysis and flow cytometry. In co-culture experiments, allogeneic DCs and macrophages were used.
Co-culture migration assays
PDL cells were cultured in 24-transwell plates (Costar; Corning, NY, USA) to confluence and then washed with PBS to remove serum. For activation, PDL cells were incubated with or without cytokines (IL-1β, TNF-α, HMGB1) for 24 h without serum. Subsequently, PDL cells were washed with PBS to remove cytokines and incubated with serum-free medium. Transwell inserts of 5.0 µm pore size (6.5 mm diameter, Polycarbonate Membrane; Corning) were placed into the wells and filled with serum-free medium containing 4 × 105 iDCs/M0 previously labeled with CFSE. As negative control, iDCs or M0 were cultured alone under the same conditions. As positive control for DC migration, iDCs alone were cultured in wells containing 0.1 µg/ml macrophage inflammatory protein (MIP) 1α in the lower chamber of the transwell. As a positive control for macrophage migration, M0 alone were cultured in wells containing 10% FCS in the lower chamber of the transwell.
After 18 h incubation time, iDCs/M0 that had migrated into the lower chamber were harvested and cell numbers were determined by flow cytometry detecting green-fluorescence (FITC) within a time-frame of 60 s using a BD FACS Canto Flow Cytometer (BD Biosciences, San Jose, CA, USA).
Co-culture maturation assays
PDL cells were cultured in 24-well plates (Greiner Bio-One, Kremsmünster, Austria) to confluence and then washed to remove serum. For activation, PDL cells were incubated with or without cytokines (IL-1β, TNF-α, HMGB1) for 24 h without serum. Subsequently, PDL cells were washed to remove cytokines and incubated with serum-free medium containing 4 × 105 iDCs/M0 for 48 h to allow direct cell–cell contact.
In order to investigate the effects induced by PDL cells in the absence of direct cell–cell contact, PDL cells were incubated with or without cytokines (IL-1β, TNF-α, HMGB1) for 24 h without serum. Subsequently, PDL cells were washed to remove cytokines and incubated with serum-free medium. After 1 h, supernatants were collected, transferred to fresh wells containing 4 × 105 iDCs/M0 and incubated for 48 h.
As a negative control for both experimental setups, iDCs/M0 were cultured alone. To induce matured mDC, iDCs alone were cultured with 1 µg/ml LPS (from Escherichia coli 055:B5; Sigma-Aldrich, St Louis, MO, USA).
Macrophage polarization was obtained by culturing M0 alone in medium supplemented with 5% FCS, 100 ng/ml LPS and 20 ng/ml IFN-γ for polarization towards a M1 type or in medium supplemented with 5% FCS and 20 ng/ml IL-4 for polarization towards a M2 type.
DCs/macrophages were then trypsinized with 2mM PBS/EDTA and prepared for FACS by labeling with fluorescent Abs. DCs were analyzed for CD11c, CD1a, CD83 and CD86, and macrophages for CD14, CD23, CD163 and CD64.
Phagocytosis assays
As phagocytosis by DCs and macrophages is essential for subsequent induction of immunity, we investigated the influence of resident PDL cells on the phagocytic capacities of these immune cells with regard to P. gingivalis.
DCs (mDCs/iDCs/co-cultured with pre- or unstimulated PDL cells) or macrophages (M0/M1/M2/co-cultured with pre- or unstimulated PDL cells) were incubated for 45 min at 37 °C with pHrodo™ labeled P. gingivalis. The ratio of DCs or macrophages:bacterial counts was 1:20. Then, cells were washed and immediately analyzed by FACS. Cells incubated at 4 °C were used as negative control.
Statistical analysis
Statistical evaluation was performed using one-way ANOVA and Tukey post-hoc test for detection of statistically significant differences between paired observations. Values were calculated for n = 6 and are expressed as mean ± SEM. P < 0.05 was considered statistically significant. Analytic tests were performed using Grahpad Prism Software (version 4, MacKiev Software).
Results
Human PDL cells stimulate the migratory capacity of DCs and macrophages
Both DCs (P = 0.0001) and macrophages (P = 0.0002) were significantly activated by PDL cells in their migratory capacity, as shown in Figure 1A–B.
Migratory response of DCs and macrophages upon co-culture with PDL cells. CFSE labeled iDCs (A) and M0 (B), respectively, were co-cultured with PDL cells previously incubated for 24 h with or without IL-1β (5 ng/ml), TNF-α (5 ng/ml), HMGB1 (100 ng/ml) in transwell systems with inserts of 5.0 µm pore size. After 18 h, DC/macrophage migration was counted by flow cytometry. MIP1α (0.1 µg/ml) was used as a positive control for DC migration. 10% FCS was used as positive control for macrophage migration. The values show the mean ± SEM (n = 6). P < 0.05. (A) *: statistically significant compared to ‘control’, #: statistically significant compared to ‘PDL IL-1β’ (A). *: is statistically significant compared to ‘control’, #: statistically significant compared to ‘PDL’ (B).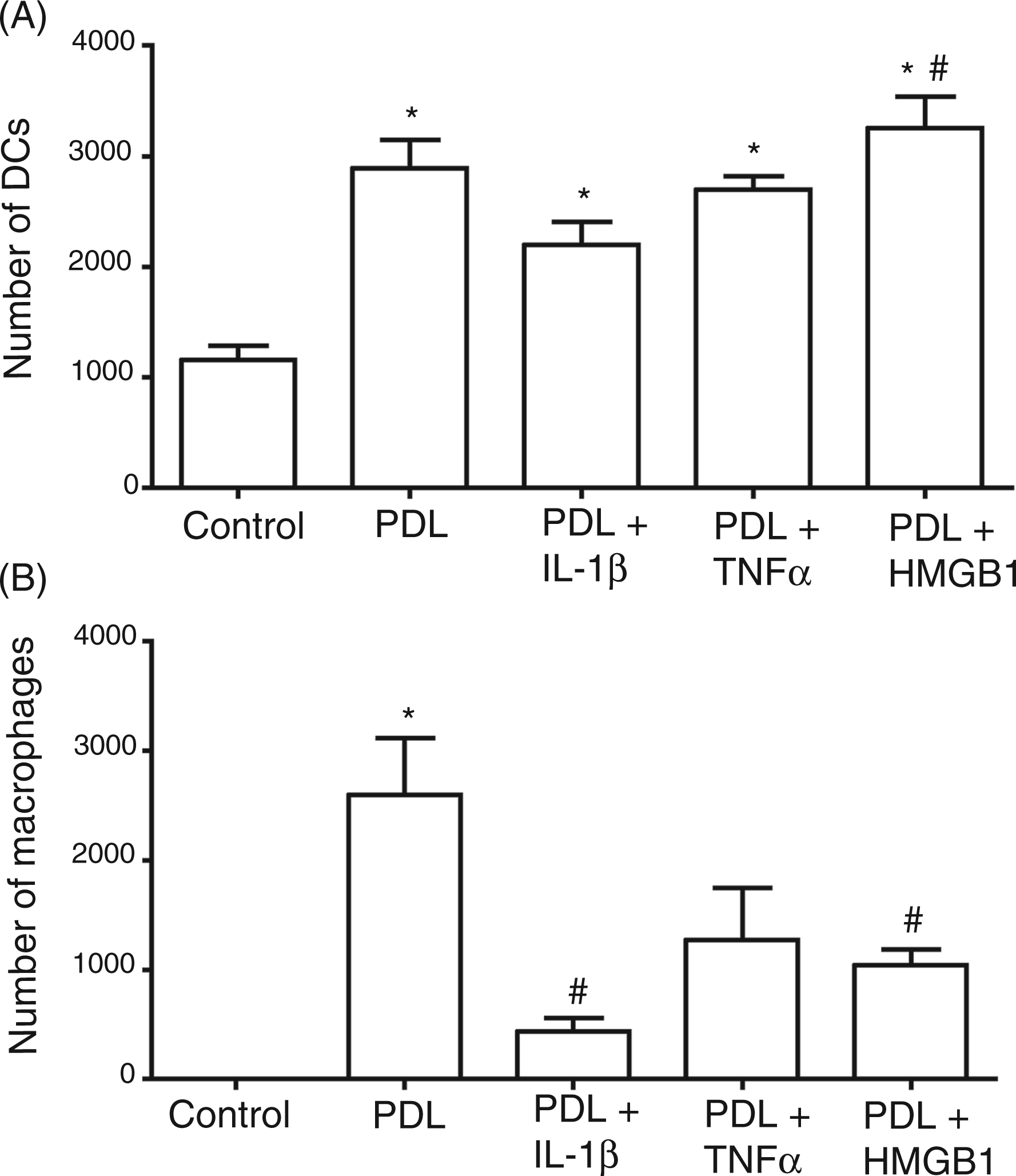
Co-culture experiments increased the migratory response of DCs, which was most pronounced in co-cultures with PDL cells receiving HMGB1 pretreatment with 3300 ± 300 migrating cells. Statistically significant differences (P < 0.05) could be noted between PDL cells pre-stimulated with IL-1β, which evoked the weakest effects with 2200 ± 200 migrating cells, and PDL cells prestimulated with HMGB1.
Macrophages did not display any migration when cultured alone, but showed a migratory response when co-cultured with PDL cells without inflammatory priming (P = 0.001). Inflammatory challenge of PDL cells decreased these effects with statistically significant differences between macrophage migration of 2600 ± 500 cells upon co-culture with unstimulated PDL cells and those prestimulated with IL-1β (400 ± 100; P = 0.01) and with HMGB1 (1000 ± 100 migrating cells; P = 0.05).
Co-culture with PDL cells promotes the maturation of DCs and macrophages
Significantly increased expression of the DC maturation markers CD83 (P = 0.0001) and CD86 (P = 0.0017) could be noted when co-culturing iDCs with PDL cells (Figure 2). CD83 was significantly enhanced by the presence of PDL cells up to 23% ± 2. In contrast, upregulation of CD86 was exclusively significant when iDCs were co-cultured with prestimulated PDL cells (39% ± 1).
Impact of PDL cells on DC maturation. iDCs were co-cultured with PDL cells previously incubated for 24 h with or without IL-1β (5 ng/ml), TNF-α (5 ng/ml), HMGB1 (100 ng/ml). After 48 h, DCs were analyzed by flow cytometry for CD83 and CD86. As a negative control, iDCs were cultured alone. As a positive control, iDCs were cultured alone with 1 µg/ml LPS to induce mDCs. The values show the mean ± SEM (n = 6). P < 0.05. *: statistically significant compared to CD83 on iDCs, #: statistically significant compared to CD86 on iDCs.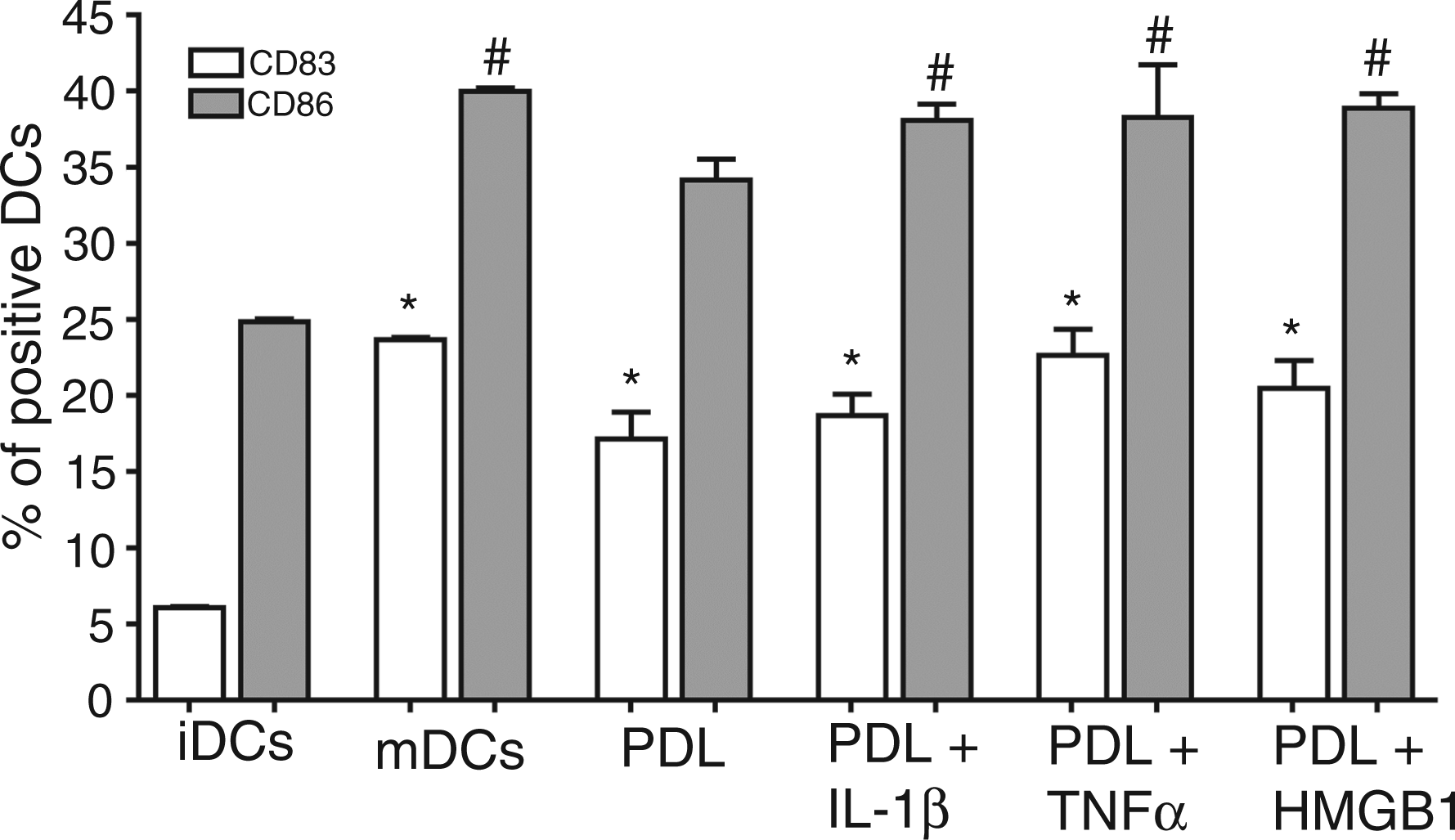
Interaction of macrophages with PDL cells induced a significant downregulation of the M1 maturation marker CD64 (P < 0.0001) down to 29% ± 3, even below the expression level found in the M2 induced macrophages (46% ± 0). The effects were also statistically significant when compared to the M1-induced macrophages (73% ± 0). Furthermore, no significant differences between co-culturing with untreated or prestimulated PDL cells could be noted (Figure 3A). In contrast, both M2 markers CD23 (P = 0.0005) and CD163 (P = 0.0023) were significantly upregulated upon interaction with PDL cells. In comparison to both M0 and M1 induced macrophages (1% ± 0), CD23 (Figure 3B) was significantly upregulated up to 41% ± 6 upon co-culturing with PDL cells; the extent was equivalent to the expression level in M2 induced macrophages. CD163 (Figure 3C) was significantly increased after co-culturing with HMGB1 prestimulated (21% ± 2) or unstimulated PDL cells (17% ± 2).
Impact of PDL cells on macrophage maturation. M0 were co-cultured with PDL cells previously incubated for 24 h with or without IL-1β (5 ng/ml), TNF-α (5 ng/ml) and HMGB1 (100 ng/ml). After 48 h, macrophages were analyzed by flow cytometry for the M1 marker CD64 (A), the M2 marker CD23 (B) and the M2 marker CD163 (C). As a negative control, M0 were cultured alone. As a positive control, M0 were cultured with 5% FCS, 100 ng/ml LPS and 20 ng/ml IFN-γ for polarization towards a M1 type or with 5% FCS and 20 ng/ml IL-4 for polarization towards a M2 type. The values show the mean ± SEM (n = 6). P < 0.05. *: statistically significant compared with M0, #: statistically significant compared to M1.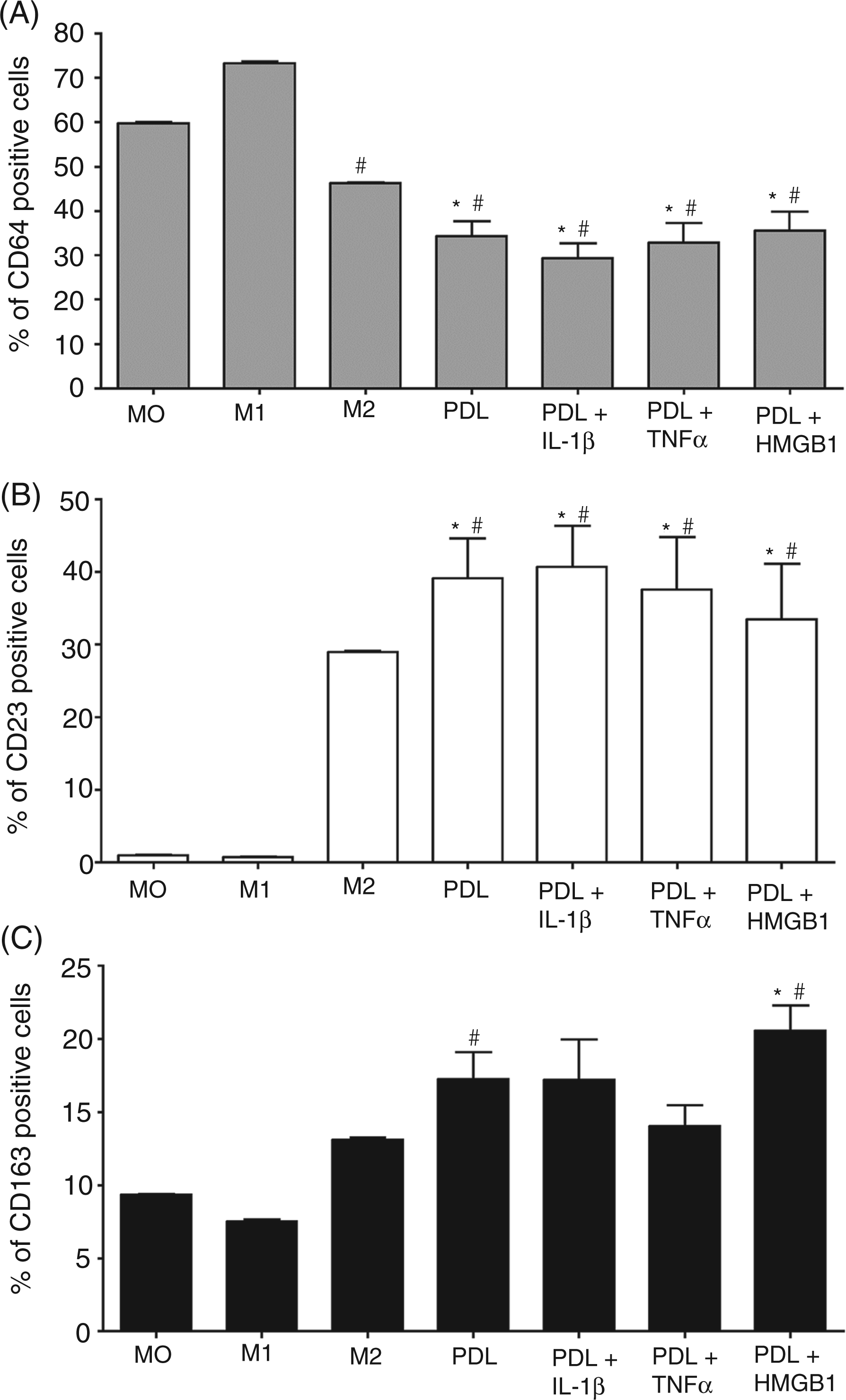
Macrophage and DC differentiation was also significantly (P < 0.0001) modulated by soluble factors released by PDL cells. CD83 was induced to the same extent as co-culturing with PDL cells, with statistical significance for IL-1β and HMGB1 prestimulated cells. The rise in CD86 expression was about 24% less in relation to co-culture with PDL cells, with statistical significance for unstimulated and IL-1β prestimulated PDL cells. CD64 upregulation was not significantly different between the expression levels induced by co-culture and the ones induced by isolated soluble factors. In contrast, CD23 was induced about 86% less and CD163 about 76% less by isolated soluble factors. Figure 4 is an exemplary illustration of these effects using CD163 as an example to compare the effects induced by isolated soluble factors and by co-culture.
Comparison of the impact of PDL cells and of their isolated soluble factors on macrophage maturation. PDL cells were incubated with or without IL-1β (5 ng/ml), TNF-α (5 ng/ml), HMGB1 (100 ng/ml) for 24 h, subsequently washed to remove cytokines and incubated with serum-free medium. After 1 h, supernatants were transferred to fresh wells and incubated for 48 h with iDCs/M0. The values show the mean ± SEM (n = 6). P < 0.05. *: statistically significant.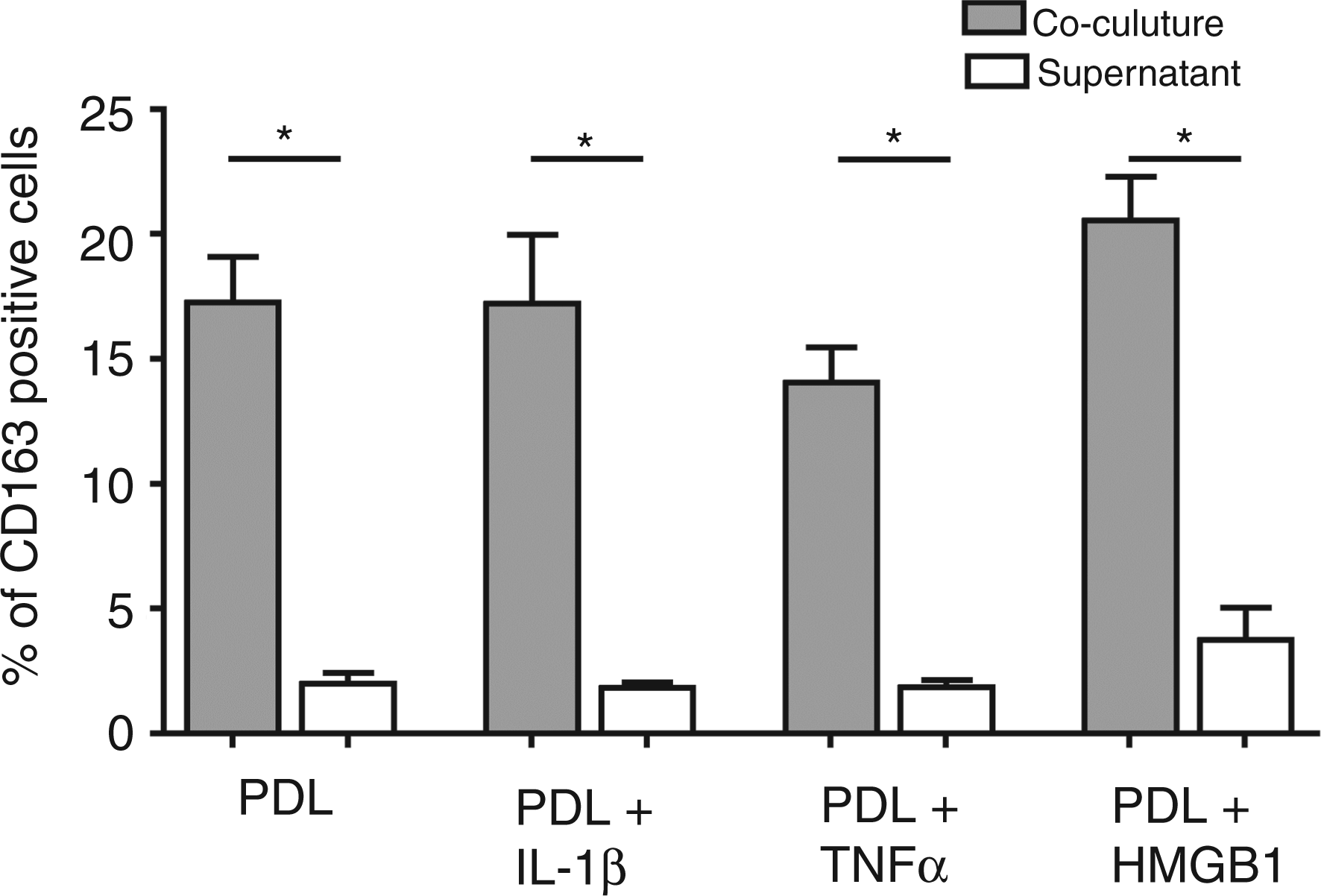
PDL cells impact the phagocytic ability of professional phagocytes
In our experiments, co-culturing of DCs with PDL cells significantly upregulated the ingestion of P. gingivalis up to 99% in the mean in comparison to both iDCs (98% ± 0) and mDCs (97% ± 0). Prestimulation of PDL cells did not have any additional effect (Figure 5A). The same pattern was obvious in the macrophage experiments, where co-culturing with PDL cells also resulted in significantly enhanced phagocytosis of P. gingivalis up to 97% ± 1 compared to M0 (72% ± 0), M1-induced (82% ± 0) and M2-induced macrophages (88% ± 0). Unlike DCs, macrophage activation was differentially modulated by prestimulation of PDL cells, as treatment of PDL cells with HMGB1 induced a significantly lower phagocytic activity in macrophages than PDL cells challenged with TNF-α, IL-1β or without any pretreatment (Figure 5B).
Impact of PDL cells on the phagocytosis of P. gingivalis by DCs and macrophages. iDCs (A) and M0 (B), respectively, were co-cultured with PDL cells previously incubated for 24 h with or without IL-1β (5 ng/ml), TNF-α (5 ng/ml), HMGB1 (100 ng/ml). After 48 h, DCs/macrophages were removed and incubated for 45 min with pHrodo™ labeled P. gingivalis (ratio 1:20 of DCs/macrophages:bacterial counts) and subsequently analyzed by flow cytometry. The values show the mean ± SEM (n = 6). P < 0.05. *: statistically significant compared to iDCs, #: statistically significant compared to mDCs (A). *: statistically significant compared to M0, #: statistically significant compared to M1, ×: statistically significant compared to M2, +: statistically significant compared to HMGB1 (B).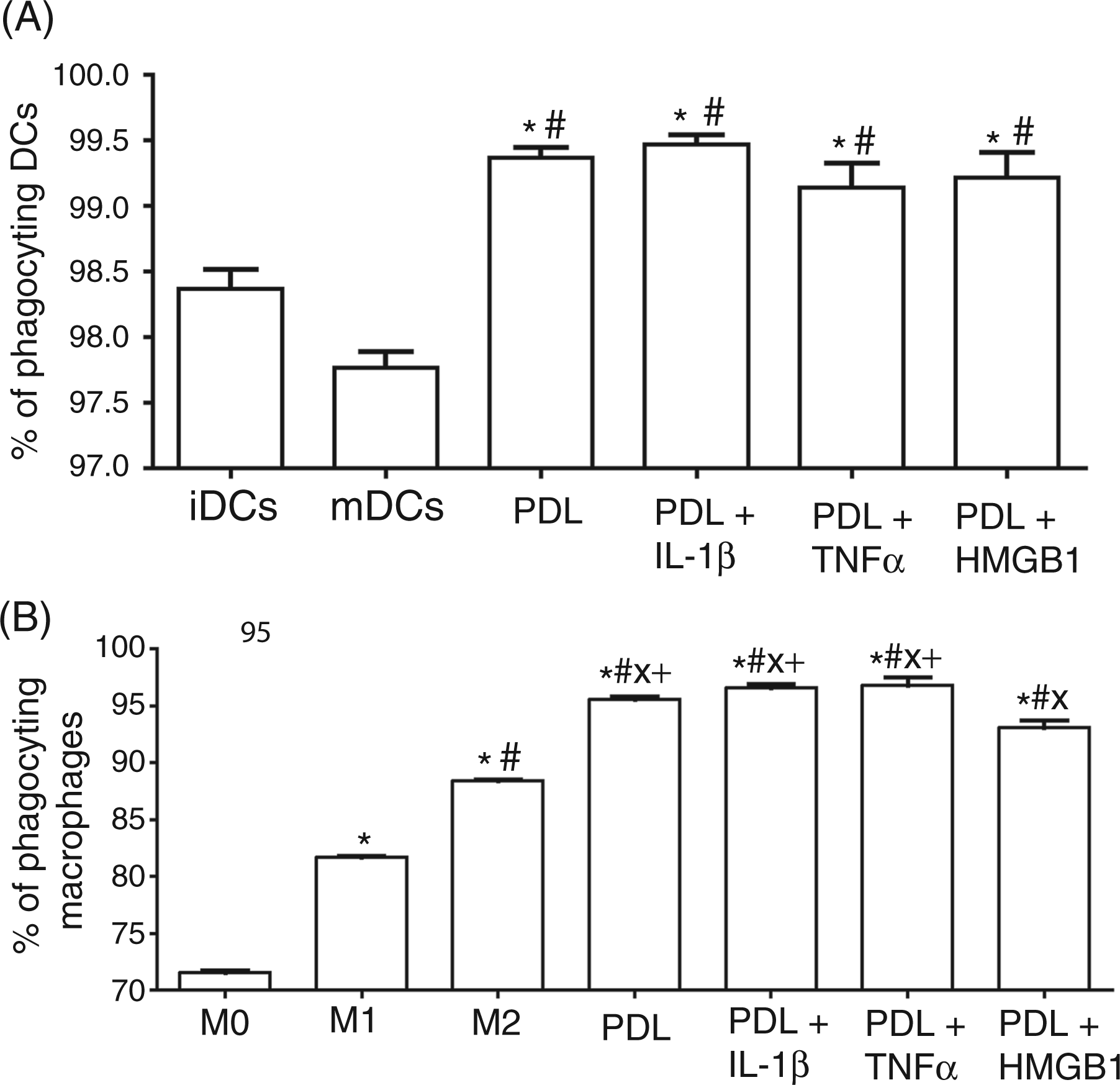
Discussion
Our experiments revealed that PDL cells interact with immune cells under both non-inflammatory and inflammatory conditions by enhancing their migration, by facilitating their phagocytic capacities and by affecting their phenotypic maturation both through cell–cell contact and through released soluble mediators.
This study was designed to investigate the influence of PDL cells on the local innate immune responses in the periodontium. Phagocytic cells control the balance between pro- and anti-inflammatory reactions, but a disruption of this equilibrium in immune defense may lead to host-mediated tissue damage. At this time, little is known about the interaction of innate immune cells and the cellular microenvironment these cells encounter during host defense.
Our experiments revealed that PDL cells are not only structural elements of the periodontium, but actively influence innate immune responses by interaction with both DCs and macrophages.
PDL cells did not only promote the migration of these innate immune cells, but also induced their phenotypic maturation and modulated their phagocytic activity both under non-inflammatory and inflammatory conditions.
We could demonstrate that the migration of phagocytes, which is an essential step in the initiation of an immune response, is enhanced by PDL cells through their release of soluble factors, which seem to provide a functionally important microenvironment for DC and macrophage activation. The migration of DCs was significantly enhanced by the presence of PDL cells in general, but HMGB1-activated PDL cells provoked the most pronounced effects. Saalbach et al. studied whether interaction of DCs with dermal fibroblasts favors the migration of DCs and found that unstimulated dermal fibroblasts failed to induce DC migration, whereas inflammatory-activated fibroblasts stimulated their migration. 29 Studies on DC recruitment by neutrophils reported a potent chemotactic function of the chemokines CC chemokine ligand (CCL) 3, CCL4, CCL5 and CCL20, which are (with the exception of CCL4) also expressed in PDL cells.30–33 Inflammation and especially the properties of HMGB1 might modulate the synthesis and interaction of these chemokines and thus augment the potency of their chemotactic activity. It is known that HMGB1, which is released by necrotic or damaged cells, but also secreted by activated monocytes and macrophages, evokes inflammation as much as chemotaxis of cells to resolve tissue damage and thus also seems to occupy an important function in DC chemotaxis induced by HMGB1-activated PDL cells.34,35
With respect to macrophages, migration of these cells was only induced after co-culturing with PDL cells, which was most pronounced for unstimulated PDL cells. Recent studies revealed that S100A4, a family member of the S100 Ca2+-binding proteins, also known as fibroblast-specific protein, is not only important for the physiologic motility of macrophages, but also regulates their chemotaxis and recruitment. 36 Increased S100A4 expression was also found at sites of inflammation and in rheumatoid arthritis, particularly at sites of joint destruction. 37 This might indicate a contributory role for S100A4 in the pathogenesis of chronic auto-inflammatory diseases and could also be considered in the case of periodontitis.
Furthermore, our experiments demonstrated that PDL cells are capable of inducing phenotypic maturation of DCs and macrophages via cell–cell contact, as well as via the release of soluble mediators. In DCs, PDL cells triggered upregulation of both DC maturation markers investigated. Whereas CD83 was significantly upregulated by co-culture with PDL cells and to the same extent by their isolated soluble mediators, CD86 was induced to a lower degree by the exclusive exposure to isolated soluble factors released by PDL cells. Experiments on DC maturation induced by dermal fibroblasts also discovered an effect of both cell–cell contact and soluble mediators released by dermal fibroblasts on DC maturation. 38 For the maturation process, the authors pointed out the importance of TNF-α induction in DCs, which was exclusively affected by direct cell contact with dermal fibroblasts, but discussed that other signals apart from TNF-α must impact DC maturation, as they also observed upregulation of DC maturation molecules in physically separated co-cultures.
Additionally, our analyses revealed that PDL cells induce phenotypic maturation of macrophages with distinct oppositional patterns for the M1 and M2 characteristic macrophage markers. Both resting and inflammatory-primed PDL cells in co-culture, as much as their isolated soluble products, downregulated the M1 marker CD64 to the same extent, which resulted in expression levels even below the ones found in M0 macrophages. In contrast, upregulation of both M2 markers CD23 and CD163 could be noted upon co-culture experiments and, to a lower extent, upon exposure to isolated soluble factors by unstimulated and by inflammatory-primed PDL cells. The resulting expression levels reached the same level as in the IL-4-induced M2 macrophages.
Evidence exists that differential modulation of the chemokine system affects macrophage function with respect to resistance or immunoregulation, host defense and tissue repair. M1 macrophages develop microbicidal and anti-tumoral properties, whereas M2 macrophages have immunomodulatory properties to control inflammation and are involved in tissue remodeling.39,40 Consequently, the differentiation of macrophages in tissues seems to be characterized by a drift towards an immunomodulatory phenotype evoked by resident periodontal cells.
In order to investigate whether PDL cells support the transition from the innate to an adaptive immune response, we analyzed whether PDL cells influence the phagocytic capacities of DCs and macrophages as professional APCs, which is an essential step preceding Ag presentation to lymphocytes. As inflammation induced by periodontal pathogens stimulates both innate and adaptive immune responses, but not necessarily leading to tissue destruction, 41 we examined the impact of resident PDL cells on the phagocytosis pattern of DCs and macrophages towards P. gingivalis. The gram-negative anaerobic bacterium represents one of the best described bacterial species to be associated with periodontitis and is, besides Treponema denticola and Tannerella forsythia, strongly related to advanced periodontal lesions. 42 Our experiments with avital P. gingivalis showed that PDL cells stimulated phagocytosis in professional phagocytes above the levels found in iDCs or each of the different macrophage phenotypes. As phagocytosis is pronounced differently in these cell types depending on their degree of maturation, PDL cells seem to enhance this host defense mechanism against bacterial insults and thus may indirectly impact periodontal tissue destruction.
Finally, it has to be recognized that we used a classification of macrophages in the ‘classical activation state’ (M1) and ‘alternative activation state’ (M2) in our in vitro experiments. However, despite this classification, the extent of heterogeneity and plasticity in the system of mononuclear phagocytes is still an ongoing matter of debate. Thus, it is still unclear whether after differentiation and activation macrophage fate is determined or whether it is permanently susceptible to change. Furthermore, it is unclear whether distinct activation states exist in vivo or whether macrophages show a broad range of phenotypes instead.19,43 In addition, it is discussed whether there is a clear-cut distinction between DCs and macrophages or whether a continuum of cellular phenotypes of cells of the mononuclear phagocyte system exists.44,45 Thus, it has to be assumed that in vivo, an inflammatory environment leads to the exposure of macrophages to multiple stimuli with complex phenotypic consequences.
Taken together, the results of our study show that PDL cells actively impact the local innate immune response in periodontal tissues by chemotaxis of both DCs and macrophages, by modulating their maturation and by increasing the phagocytic capacities of these professional APCs. Nevertheless, it has to be considered that specific environmental and host-specific factors are likely to impact these interactions in vivo and thus determine the individual local immune response and the susceptibility to periodontal disease.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Acknowledgements
The authors would like to thank Dr M. Kebschull (KFO 208, Department of Periodontology, Operative and Preventive Dentistry, University of Bonn) for kindly providing the P. gingivalis 381 strain, and S. Hegenbarth (Institute of Experimental Immunology, University of Bonn) and N. Gallala (Department of Oncology and Hematology, University of Bonn) for technical support.
Conflicts of interest
The authors have no conflicts of interest to declare.