
Abstract
Infection with a variety of bacterial pathogens results in hematopoietic stem and progenitor cell (HSPC) mobilization. The mechanism and kinetics of HSPC mobilization during infection are largely unknown. Previously, we found altered HSPC activity in bone marrow, spleen and blood during infection with Anaplasma phagocytophilum, the agent of granulocytic anaplasmosis. We hypothesized that altered CXCL12/CXCR4 signaling, a central pathway for HSPC homing to, and retention within, the bone marrow, plays a role in infection-induced alterations in HSPC number and trafficking. Mice were infected with A. phagocytophilum. Lineage-cKit+ HSPCs were enumerated and proliferation determined. CXCL12 and CXCR4 mRNA were quantified along with CXCL12 protein, and CXCR4 surface, intracellular and total protein expression in HSPCs was determined. Increased bone marrow proliferation of HSPCs began at 2 d post-infection followed by HSPC mobilization and splenic homing. Proliferation of resident HSPCs contributed to increased splenic HSPC numbers. Bone marrow CXCL12 mRNA and protein levels were decreased at 4–8 d post-infection concurrent with HSPC mobilization. CXCR4 protein parameters were decreased in bone marrow HSPCs throughout 2–6 d post-infection. Reduction of CXCL12/CXCR4 signaling simultaneously occurs with HSPC mobilization from bone marrow. Findings suggest that deranged CXCL12/CXCR4 signaling plays a causal role in HSPC mobilization during acute A. phagocytophilum infection.
Introduction
The CXCL12/CXCR4 signaling axis controls the homing and mobilization of hematopoietic stem and progenitor cells (HSPCs) and mature leukocytes.1–3 Previous work on this signaling pathway has primarily focused on its role in transplantation and stem cell biology, as HSPC mobilization can be initiated clinically using a selective antagonist to this ligand/receptor interaction.4,5 More recently, a role has emerged for CXCL12 signaling in neutrophil trafficking to and from bone marrow, both in homeostasis and during inflammation.6–8 Chemokine signaling in leukocyte trafficking regulates the availability of immune effector cells at sites of inflammation and infection.
Alterations in CXCL12 signaling during bacterial infection may also be critical for controlling the trafficking of HSPCs and influencing infection outcome. Regulation of hematopoiesis via altered HSPC kinetics can increase production of multiple hemic cell lineages to replace and/or augment cell populations consumed in responding to infection. Any serious derangement of hematopoiesis during bacterial infection may reduce the availability of immune effector cells and influence disease pathology. To date, there is no mechanistic, kinetic evaluation of how a bacterial infection regulates CXCL12/CXCR4 signaling in vivo.
Anaplasma phagocytophilum is a Gram-negative, LPS-negative, obligate intracellular bacterium that resides primarily within circulating granulocytes.9,10 Anaplasma phagocytophylum is the etiologic agent of granulocytic anaplasmosis, the second most common human tick-borne disease in the USA. 11 Infection with A. phagocytophilum typically results in multiple cytopenias (thrombocytopenia, leukopenia and anemia) in natural disease and in animal models of infection.12–17 Cytopenias and bone marrow dysfunction contribute to serious clinical sequelae of infection, including fatalities as a result of immunosuppression and secondary infections.12,18 Pathogen effects on host granulocytes have been well-studied.19,20 Experimentally, A. phagocytophilum infection results in altered neutrophil trafficking, increased neutrophil surface integrin expression and decreased neutrophil apoptosis with resultant effects on pathogen clearance.16,21 The impact of infection on other hemic cell lineages and hematopoiesis are poorly understood. Anaplasma phagocytophylum infection provides a useful model for investigating hematopoietic alterations arising in infections with diverse non-LPS and obligate intracellular pathogens.
Bone marrow CXCL12 mRNA expression is downregulated during acute A. phagocytophilum infection. 22 This downregulation occurs in concert with altered HSPC colony-forming activity in the bone marrow, spleen and peripheral blood along with shifts in lineage-committed cells. HSPCs express CXCR4, the primary CXCL12 receptor. HSPCs respond to directional cues created by a CXCL12 concentration gradient by mobilizing when bone marrow CXCL12 levels decrease and homing toward an increasing chemokine gradient. 23 Downregulated CXCL12 signaling may therefore actuate some of the hemic cell alterations in A. phagocytophilum infection.
The objectives of this study were to quantify HSPC proliferation, mobilization and trafficking to spleen and blood during acute A. phagocytophilum infection and to fully characterize the regulation of CXCL12/CXCR4 signaling during infection. We hypothesized that acute infection would result in HSPC proliferation, disruption of bone marrow CXCL12/CXCR4 interactions and mobilization of HSPCs from bone marrow to the periphery. We found that A. phagocytophilum infection resulted in rapidly increased bone marrow HSPC proliferative activity followed by mobilization of HSPCs into peripheral blood. Proliferative activity also increased in splenic HSPCs, which contributed slightly to total mobilized HSPCs in blood. Our data implicate kinetic regulation of CXCL12/CXCR4 signaling as the mechanism of HSPC mobilization. Simultaneous downregulation of bone marrow CXCL12 mRNA transcription and protein production occurred along with reduction in both surface and intracellular bone marrow CXCR4 expression. Infection with this non-LPS pathogen altered CXCL12/CXCR4 signaling and increased HSPC proliferation and mobilization during the innate immune response to infection.
Methods
Mice and in vivo experimental infection
Mouse experiments were performed exactly as previously described. 21 Female, 5- to 7-wk-old, C3H/HeN (C3H) and C3H/HeN SCID (C3H-SCID) mice werepurchased from Harlan Sprague-Dawley (Indianapolis, IN, USA) and Taconic (Hudson, NY, USA). For some experiments, C3H mice were splenectomized by Taconic according to their protocol and specifications. Experimental C3H mice were inoculated via i.p. injection with 200 µl of pooled whole blood from infected C3H-SCID mice. 24 Anaplasma phagocytophylum strain NCH-1 was utilized for all infections. In some experiments, mice were injected i.p. 2 h before euthanasia with 100 μl of 10 mg/ml BrdU solution (APC BrdU Flow Kit, BD Biosciences, San Jose, CA, USA). Mice were maintained according to approved institutional animal use and care protocols. Mice were euthanized with CO2 on days 0 (control), 2, 4, 6 and 8 post-infection.
Tissue collection and processing
Blood, spleen and bone marrow were collected at necropsy. Blood was collected into EDTA by cardiocentesis. Whole blood was used for quantitative PCR (qPCR) to detect A. phagocytophilum p44 DNA, complete blood cell counts (CBCs), differential leukocyte counts and flow cytometry.
16
CBCs were performed on an automated analyzer (Beckman Coulter AcT Hematology Analyzer; Beckman Coulter, Holbrook, NY, USA) within 4 h of blood collection. For flow cytometry, red blood cells were lysed using ammonium chloride (Stem Cell Technologies, Vancouver, BC, Canada) and samples washed twice in PBS before labeling. Remaining whole blood was centrifuged at 1500 g for 5 min and plasma was banked for ELISA analysis. The spleen was removed and weighed. A section of spleen was placed in 10% formalin for immunohistochemistry. A single cell suspension was made from the remaining spleen for flow cytometic analysis, RT-PCR, cell sorting and/or culture for determination of secreted CXCL12. In brief, the spleen was gently crushed through a 70 µm nylon sterile strainer (BD Biosciences) and resuspended in Iscove's Modified Dulbecco’s Media (IMDM; Invitrogen, Carlsbad, CA, USA) + 2% fetal bovine serum (FBS). Splenic cells were also banked at −80 oC for RT-PCR evaluation. The sternum was removed and placed in 10% formalin for immunohistochemistry. Bone marrow was sterilely harvested from femurs and tibias. In brief, intact bones with musculature removed were disinfected in 70% ethanol for 2 min and washed with sterile PBS. Bone marrow was flushed from tibias and femurs with IMDM + 2% FBS + 5 m
DNA extraction and quantitative PCR
To verify infection, DNA was extracted from 25 µl of whole blood from each mouse using the DNeasy Tissue Kit (Qiagen, Valencia, CA, USA) according to manufacturer's instructions and exactly as previously described.21,25 Amplification of DNA, data acquisition and data analysis were performed in a 7300 Real-Time system (Applied Biosystems, Foster City, CA, USA) exactly as previously described.16,26
RNA extraction and RT-PCR
Total RNA was isolated using the RNeasy mini kit (Qiagen), reverse transcribed to cDNA using the Quantitect Reverse Transcription kit (Qiagen) and amplified using Taqman Universal PCR Mastermix (Applied Biosystems). Inventoried primer/probe sets (Taqman Gene Expression Assays, Applied Biosystems) were used for mouse CXCL12, CXCR4 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; housekeeping gene). Reactions were performed in duplicate or triplicate using 4–12 ng template cDNA. Quantitative PCR was performed using a 7300 Real-Time System (Applied Biosystems). Ct values were analyzed using the 2(-ddCt) method. 27
Flow cytometry and cell-sorting
Single cell suspensions of bone marrow, spleen and whole blood nucleated cells were brought to 1 × 106 cells/ml in staining buffer (PBS + 3% FBS). Cells were labeled with antibodies against CD117/c-kit (clone 2B8), CD45 (clone 30-F11) and a cocktail of lineage markers comprised of antibodies against Gr-1/Ly6C/G (clone RB6-8C5), TER119/Ly7/6 (clone TER119), CD3 (clone 17A2), and B220 (clone RA3-6B2). All Abs were obtained from BD Pharmingen (San Jose, CA, USA) except B220 (R&D Systems, Minneapolis, MN, USA). FITC- and phycoerythrin-conjugated isotype Abs were used to prepare negative controls. Cells were incubated with Ab at room temperature for 20 min, washed and resuspended in staining buffer. For BrdU analysis, cells were labeled as above with the addition of anti-BrdU Ab (APC BrdU Flow Kit, BD Biosciences). Cells were then fixed and permeabilized according to kit directions before resuspension in staining buffer
CXCL12 immunohistochemistry
Formalin-fixed tissues were embedded in paraffin then 4 μm sections were cut and mounted on glass slides. Slides were deparaffinized with xylene and rehydrated by serial incubations with decreasing concentrations of ethanol. For antigen retrieval, slides were placed in tris buffered saline (TBS) + 0.05% Tween 20 at pH 9.0 and heated in a steamer. The sections were first blocked with PeroxAbolish (Biocare Medical, Concord, CA, USA) followed by Background Sniper (Biocare Medical) then incubated with the primary Ab (mouse anti-human/mouse CXCL12 mAb (clone 79018; R&D Systems) for 45 min. MACH2 mouse HRP Polymer (Biocare Medical) was applied (30 min incubation) followed by Betazoid DAB Chromagen (8 min incubation; Biocare Medical) with a final counter stain with hematoxylin. For bone marrow evaluation, all positive cells were counted in 3 entire sternebrae in sagittal section. For splenic evaluation, positive cells were enumerated in 5–50X fields of white pulp and 5–50X fields of red pulp; results were evaluated both separately (white pulp vs red pulp) and together.
Bone marrow and spleen CXCL12 production
Bone marrow and spleen cells from infected and control mice were re-suspended in Mesencult mouse basal medium (Stem Cell Technologies) + 10% FBS and plated in a 96-well plate at 4 × 106 cells/well. Cells were incubated at 37 °C in 5% CO2 for 48 h before harvesting of supernatants. Supernatants were concentrated using Amicon Ultra 3 kD centrifugal filter units (Millipore, Billerica, MA, USA) per manufacturer instructions before measurement of CXCL12 protein.
CXCL12 enzyme-linked immunosorbent assay (ELISA)
Banked plasma and supernatants from cultured bone marrow and splenic cells were analyzed using CXCL12 Quantikine ELISA kit (R&D Systems) according to kit instructions.
Statistical analyses
Statistics were performed with a Student's t-test (Microsoft Office Excel 2003, Microsoft Corp., Bellevue, WA, USA). All tests were performed comparing data from infected mice at each time-point with data from control mice (time 0). Unless otherwise stated, data are included from three experiments, with two control and four infected mice per time-point per experiment. A P value of < 0.05 was considered significant.
Results
HSPCs are increased in bone marrow, spleen and peripheral blood during acute infection
To define HSPC kinetics during acute infection, populations of lineage (lin)-negative, cKit-positive cells were identified using flow cytometry in bone marrow, spleen and peripheral blood (Figure 1). Lin-negative cells were gated out using a cocktail of lineage markers; CD45 positivity was additionally used for gating in peripheral blood. Lin-negative cells were then evaluated for c-Kit positivity. Acute A. phagocytophilum infection resulted in increased numbers of HSPCs in bone marrow, spleen and blood. The percentage of bone marrow HSPCs increased by day 2 post-infection with significant increases at days 6 and 8 post-infection (Figure 1A, D). The percentage of splenic HSPCs was significantly increased at days 4–8 post-infection (Figure 1B, E) and the percentage of blood HSPCs increased at day 4 post-infection with a significant increase on day 6 post-infection (Figure 1C, F).
Flow cytometric quantification of HSPCs in bone marrow (BM), spleen and peripheral blood during acute infection. Lineage-negative (and CD45-positive for peripheral blood) cells were evaluated for cKit positivity; lineage-negative and cKit-positive populations are considered equivalent to HSPCs. Representative plots are shown for days 0, 2, 4, 6 and 8 post-infection (A–C). Mean values for HSPC percentage from three experiments were compared (D–F); statistically significant differences were found in bone marrow, spleen and blood at multiple timepoints (*P<0.05). SSC: side scatter.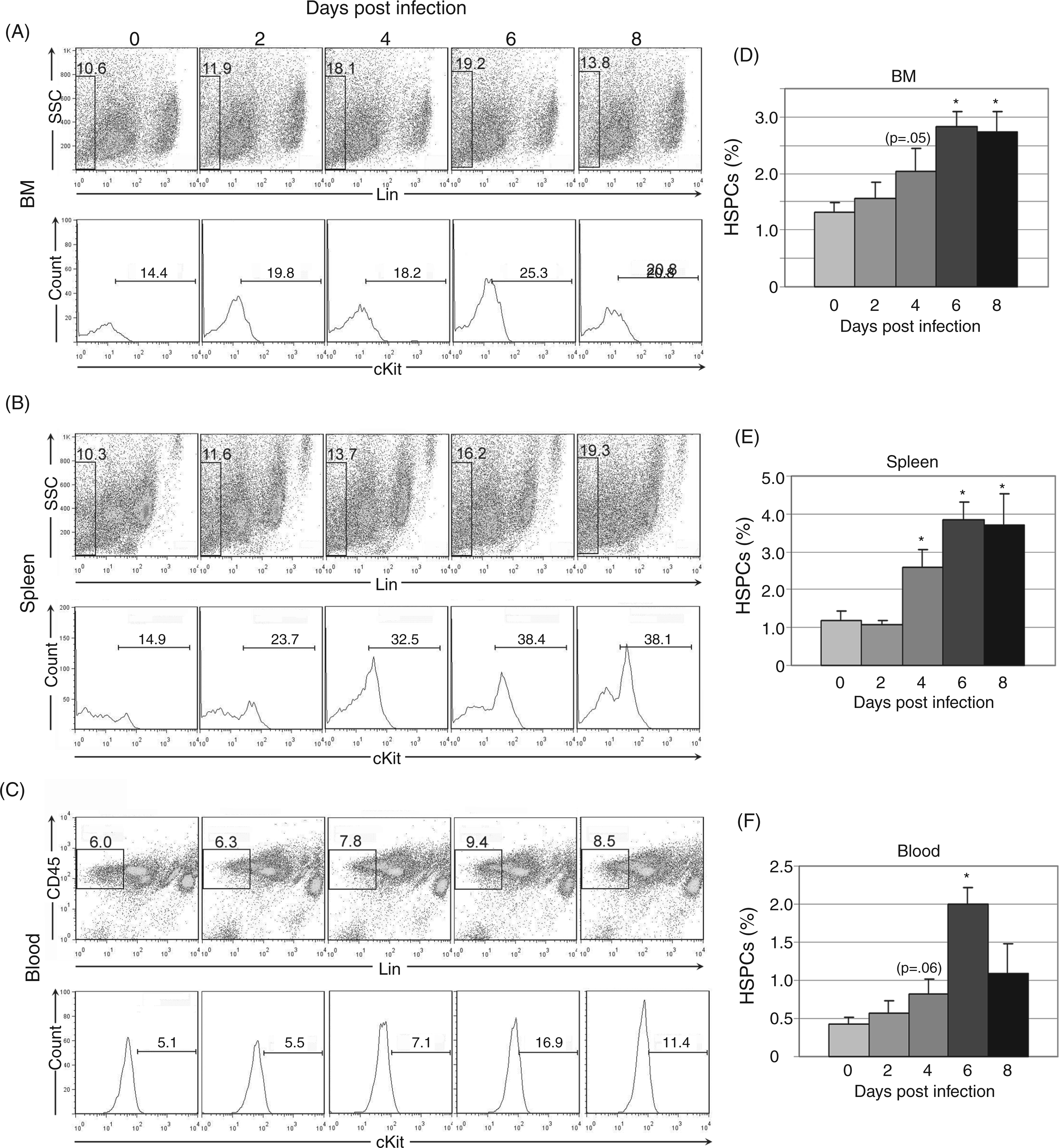
Increased numbers of HSPCs in bone marrow and spleen are due to HSPC proliferation during acute infection
To verify that HSPC numbers increased as a result of proliferation, synthetic activity was evaluated with BrdU in gated HSPC populations. Significantly increased synthetic activity in bone marrow HSPCs was found at days 2–8 post-infection (Figure 2A, C) and in splenic HSPCs at days 6–8 post-infection (Figure 2B, D). Bone marrow HSPC proliferation preceded the increase in bone marrow HSPC number, suggesting kinetic alignment of these processes. However, splenic HSPC proliferation began later than bone marrow HSPC proliferation and increased splenic HSPC number preceded splenic HSPC proliferation. These data suggest that the initial increase in splenic HSPC numbers may have been owing to an initial wave of HSPC mobilization from the bone marrow and subsequent homing to the spleen, rather than independent splenic HSPC proliferation.
Synthetic activity in bone marrow (BM) and spleen HSPCs during acute infection. Cells were first gated as in Figure 1 for lineage-negative, cKit-positive HSPC populations. BrdU positivity was then evaluated via flow cytometry. Representative plots are shown for days 0, 2, 4, 6 and 8 post-infection (A–B). Mean values for BrdU positivity in bone marrow (C) and spleen (D) HSPCs are charted; statistically significant differences were found at all infection timepoints in bone marrow HSPCs and at days 6–8 post infection in splenic HSPCs (*P<0.05).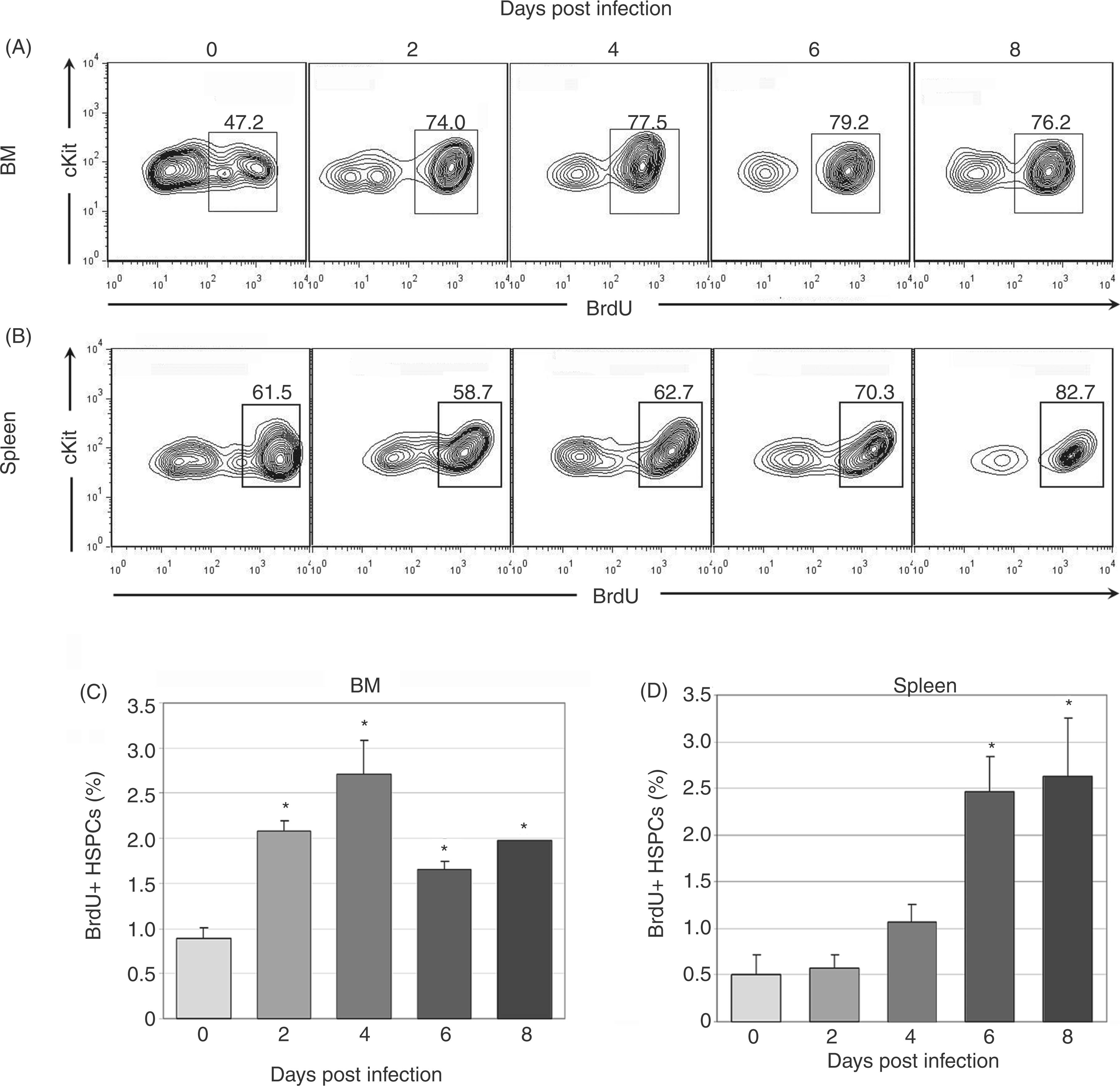
The spleen contributes to total mobilized blood HSPCs
Peripheral blood HSPC numbers increased significantly at day 6 post-infection (Figure 1E). Mobilized bone marrow HSPCs are the likely source of these cells, but the spleen may also contribute to the peripheral blood pool especially during A. phagocytophilum infection, as infection results in extramedullary hematopoiesis and reactive hyperplasia.22,28 To determine the relative contribution of splenic HSPCs to the blood pool, blood HSPC numbers were compared in splenectomized and non-splenectomized mice. Splenectomized mice had significantly fewer blood HSPCs on day 4 post-infection compared to infected mice with a spleen (Figure 3). Nonetheless, infected splenectomized mice still had a significant increase in blood HSPCs at days 4–6 post-infection, similar to the results in non-splenectomized mice shown in Figure 1. These results suggest that the spleen contributes to mobilized blood HSPCs at day 4 post-infection, but bone marrow HSPCs are sufficient, even in the absence of the spleen, to mobilize significant numbers of HSPCs during infection.
Contribution of mobilized splenic HSPCs to circulating HSPCs. Cells were gated as in Figure 1 for lineage-negative, cKit-positive HSPC populations. Compared to non-splenectomized mice (solid bars), splenectomized mice (striped bars) had significantly lower mean values for circulating HSPCs at day 4 post-infection (*P<0.05).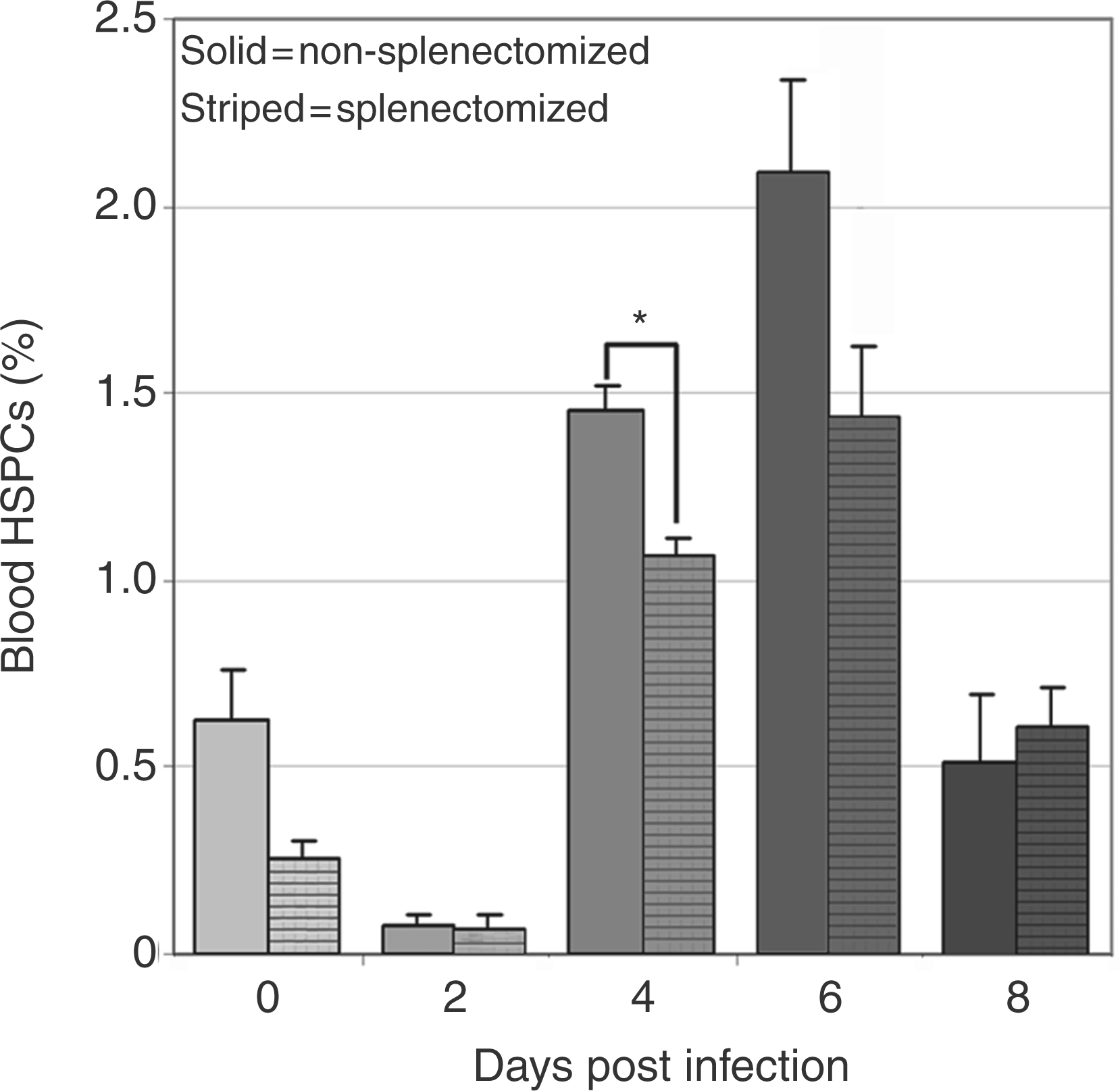
Bone marrow and plasma CXCL12 are decreased during acute infection with variable increases in splenic CXCL12
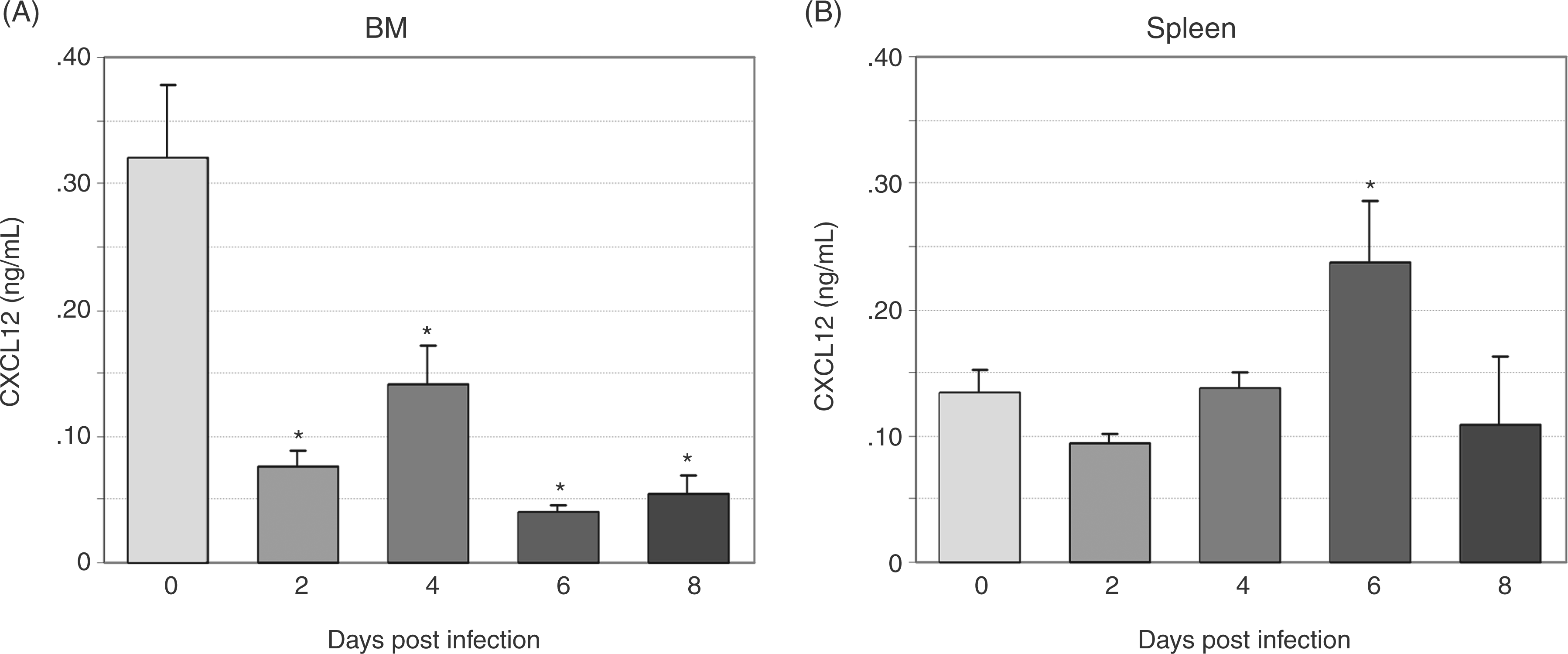
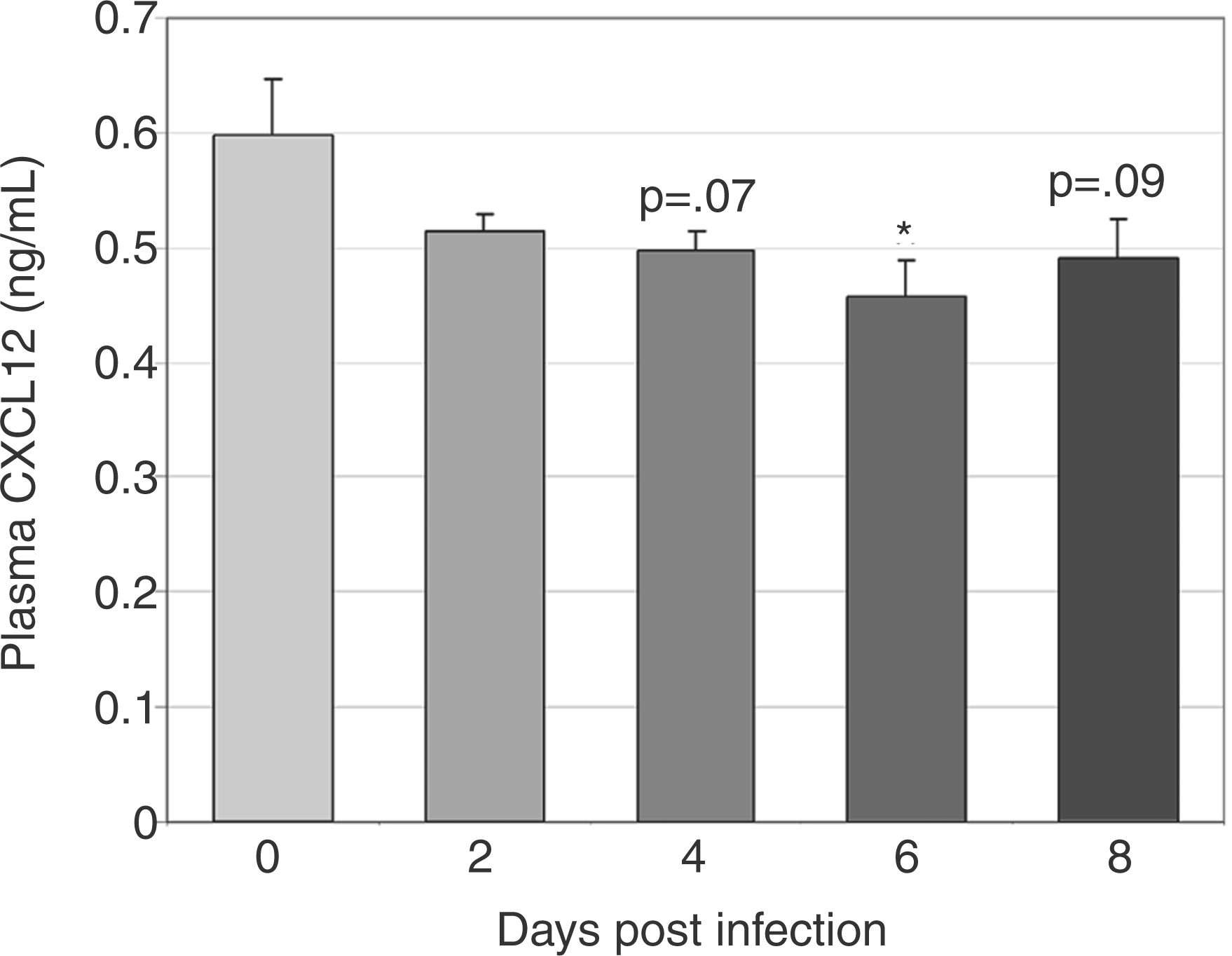
To define changes in CXCL12 protein during infection, we evaluated CXCL12 expression via immunohistochemistry in bone marrow and spleen and secreted CXCL12 via ELISA of plasma and cultured bone marrow and splenic cell supernatant. CXCL12-positive cells were significantly decreased in bone marrow at days 4 and 8 post-infection, with a trend towards a decrease at day 6 post-infection (Figure 4A, B (images at 40X) E). The percentage of splenic CXCL12-positive cells did not change with infection; however, a trend towards increased splenic CXCL12-positive cells was noted on day 8 post-infection (Figure 4C and 4D (images at 40X) F). The concentration of secreted bone marrow CXCL12 was significantly decreased at days 2–8 post-infection (Figure 5a) and the concentration of secreted splenic CXCL12 was significantly increased at day 6 post-infection (Figure 5b). Plasma CXCL12 followed a similar pattern with a significant decrease in plasma CXCL12 concentration at day 6 post-infection and a trend towards decreased plasma CXCL12 noted at days 4 and 8 post-infection (Figure 6).
Immunohistochemical evaluation of CXCL12 protein in bone marrow (BM) and spleen during acute infection. Tissues from a total of eight control mice and four infected mice per time point were evaluated. Representative images of tissue sections from control and infected (day 8) mice are shown (A–D). Mean numbers of bone marrow CXCL12-positive cells (E) were significantly decreased at days 4 and 8 post-infection, with a trend toward decrease at day 6. No significant changes in mean numbers of CXCL12-positive cells were found in splenic sections, though a trend toward an increase was seen at day 8 post-infection (F) (*P<0.05). Bone marrow (BM) and splenic CXCL12 secretion. Unfractionated bone marrow and splenic cells from a total of 12 control mice and 8 infected mice per time point were incubated in media for 48 h, and secreted CXCL12 was measured via ELISA. bone marrow CXCL12 production (A) was significantly decreased at days 2–8 post-infection. Splenic CXCL12 production (B) was significantly increased at day 6 post-infection (*P<0.05). Plasma CXCL12 levels during acute infection. CXCL12 was evaluated in plasma via ELISA. Mean values for CXCL12 from three experiments were compared; a significant decrease was found at day 6 post-infection with trends toward decrease at days 4 and 8 post-infection (*P<0.05).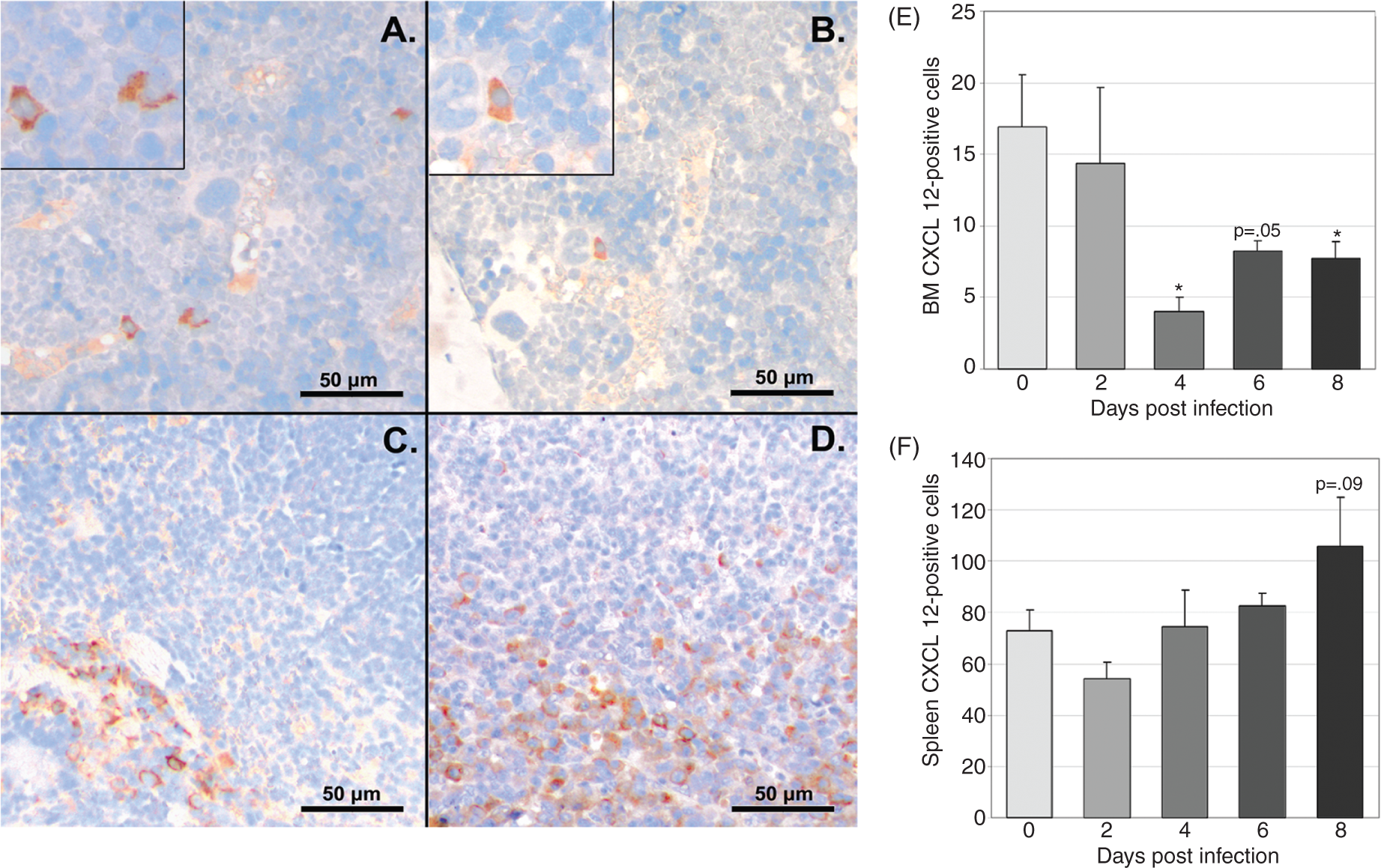
CXCL12 mRNA was quantified in bone marrow and spleen using RT-PCR. In bone marrow, CXCL12 mRNA levels were significantly decreased at days 6–8 post-infection (data not shown), consistent with our previous results. 22 In spleen, CXCL12 mRNA levels were variable and non-significant.
CXCR4 is decreased in bone marrow HSPCs and variably increased in splenic HSPCs during acute infection
Surface and total CXCR4 expression was quantified on bone marrow HSPCs throughout acute infection (Figure 7A). Mean surface CXCR4 expression onbone marrow HSPCs was significantly decreased on days 2 and 6 post-infection (Figure 7B), while mean total bone marrow HSPC CXCR4 expression (representing both surface and intracellular protein) was significantly decreased at days 2 and 4 post-infection (Figure 7b). Surface CXCR4 expression on splenic HSPCs was significantly increased at day 6 post-infection and mean total CXCR4 expression was significantly increased at day 8 post-infection (data not shown).
Flow cytometric evaluation of CXCR4 expression on bone marrow HSPCs during acute infection. Bone marrow from a total of eight control mice and four infected mice per time point was assessed. Cells were first gated as in Figure 1 for lineage-negative, cKit-positive HSPCs populations. CXCR4 positivity was then evaluated via flow cytometry. Representative plots are shown for days 0, 2, 4, 6 and 8 post-infection for surface and total CXCR4 expression (A). Mean values for percentage of CXCR4+ bone marrow HSPCs were compared (B); significant decreases were found in surface CXCR4 (solid bars) on days 2 and 6 post-infection and in total CXCR4 (striped bars) on days 2–4 post infection (*P<0.05).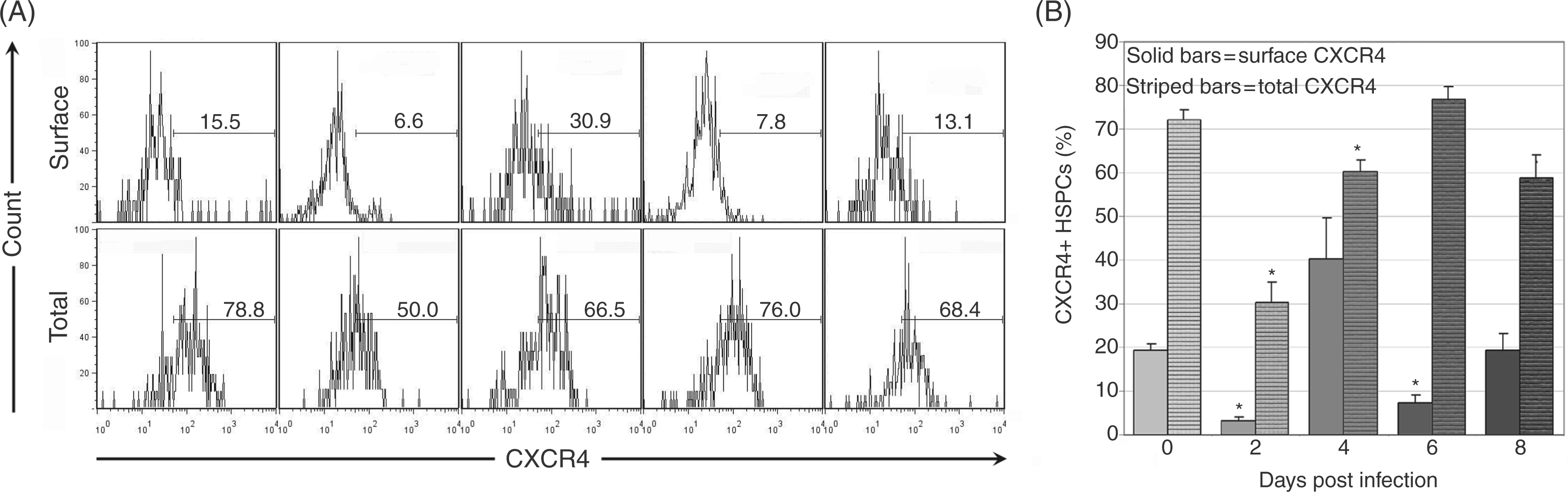
CXCR4 mRNA was quantified from sorted bone marrow (Figure 8A) and spleen (Figure 8B) HSPCs on days 4, 6 and 8 post-infection. CXCR4 mRNA did not significantly change in HSPCs from bone marrow (Figure 8C) or spleen (Figure 7D), indicating that CXCR4 regulation in HSPCs primarily occurs at a post-transcriptional level. Reciprocal changes in bone marrow and splenic HSPC receptor/ligand regulation are compatible with a model of infection-induced changes in CXCL12-CXCR4 signaling resulting in bone marrow HSPC mobilization and homing to the spleen.
Reverse transcription-PCR for CXCR4 mRNA in sorted bone marrow and spleen HSPCs. Cells were first sorted in a similar fashion as in Figure 1 for lineage-negative, cKit-positive HSPCs populations in bone marrow (A) and spleen (B). CXCR4 mRNA was then quantified via RT-PCR using the comparative Ct approach. No significant change in CXCR4 mRNA was found in bone marrow HSPCs (C). A mild, but significant, increase in fold expression of CXCR4 mRNA was found in splenic HSPCs at day 4 post-infection (D). Dpi: day post-infection.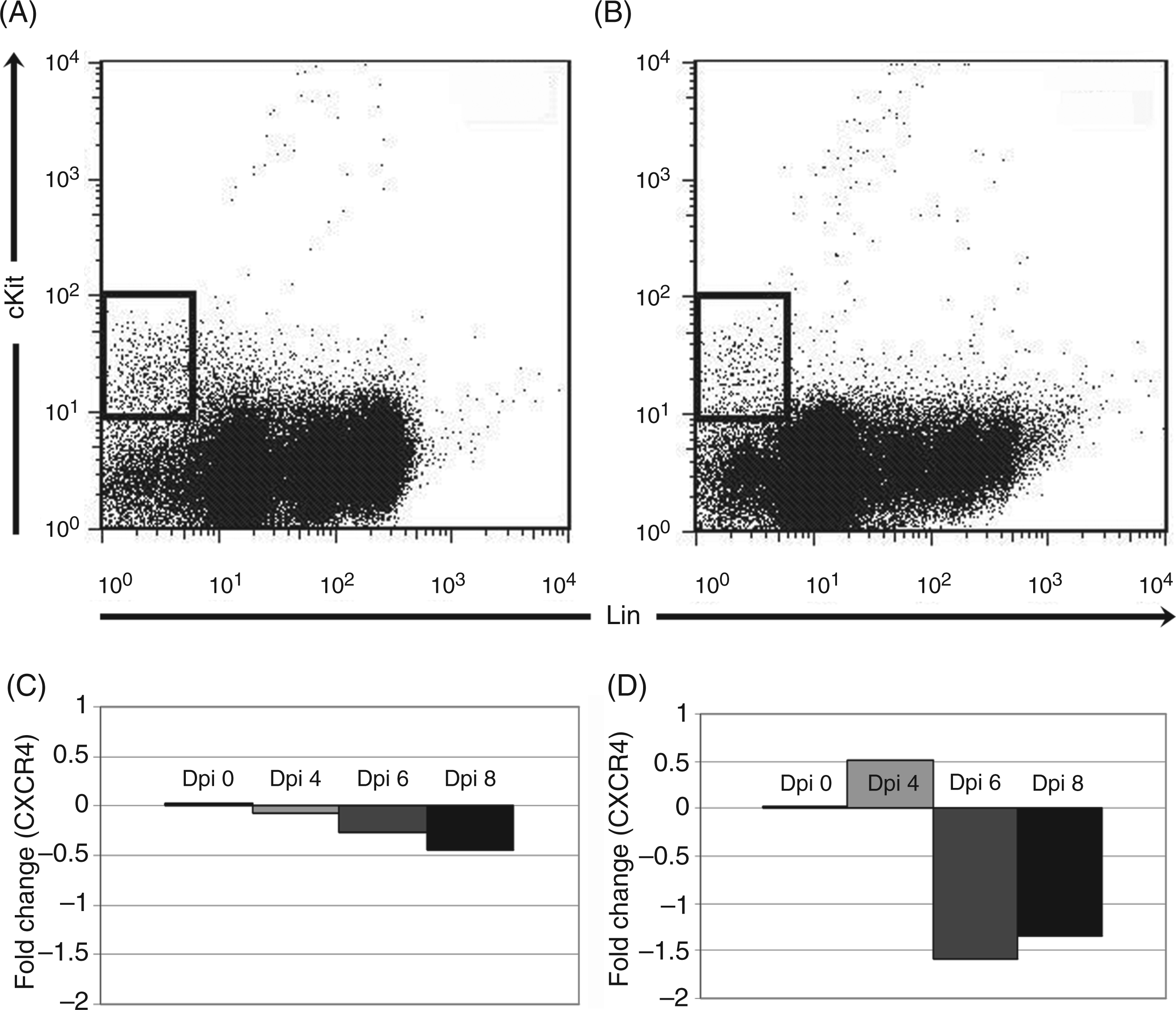
Discussion
Downregulation of CXCL12/CXCR4 signaling occurs in a murine model of infection with A. phagocytophilum, an obligate intracellular, LPS-negative and peptidoglycan layer-negative pathogen. CXCL12 is a CXC chemokine found in bone marrow stromal cells including CXCL12-abundant reticular cells, immature osteoblasts and endothelial cells.29–31 CXCR4 is expressed on HSPCs, as well as other hemic cells. 32 CXCL12/CXCR4 signaling can be modulated by inflammatory mediators. Cytokines such as TNF-α and granulocyte colony-stimulating factor (G-CSF) may suppress CXCL12 expression.33,34 Degradation of CXCL12 protein by granulocyte-derived proteases also downregulates signaling.3,35–37 Potential mediators of decreased bone marrow CXCL12 in our infection model include TNF-α and G-CSF.3,22,31 The TNF-α concentration is increased in bone marrow supernatants during infection; G-CSF alterations in A. phagocytophilum infection have not been evaluated. 22 Neutrophil proteases may also play a role in diminishing bone marrow CXCL12 protein levels.
Regulation of CXCR4 primarily occurs at the level of protein expression, as the half-life of CXCR4 mRNA is limited; however, transcriptional control does occur. Factors implicated in downregulation of CXCR4 transcription include IFN-γ, TNF-α and IL-1β. 38 The brief half-life of CXCR4 mRNA may partly explain the mild and inconsistent changes in CXCR4 via RT-PCR in sorted HSPCs noted in this study. Alternately, regulation may not occur via transcriptional control. Post-translational regulation of CXCR4 in hemic cells includes increased internalization after IFN-γ or TNF-α stimulation. 39 Taken together, our data suggest that bone marrow or systemic cytokine alterations may serve as upstream mediators of diminished CXCL12/CXCR4 signaling during acute A. phagocytophilum infection. Plasma CXCL12 levels, representing total body levels, were diminished in the later stages of acute infection, reflecting a decline in CXCL12 bone marrow production. HSPCs mobilize in response to a CXCL12 chemokine gradient, so bone marrow CXCL12 reduction likely shifts the gradient toward the periphery, resulting in HSPC mobilization. 23
Kinetic changes in CXCL12/CXCR4 in our in vivo infection model fit as a plausible mechanism of hemic cell mobilization. The role of inflammatory cytokines in the recruitment of hemic cells to a focus of infection has been well-studied. However, pathogen-induced perturbations of CXCL12/CXCR4 signaling in the bone marrow are essentially uncharacterized in vivo during bacterial infections. Viral perturbations of this pathway are better described, in part because CXCR4 is a co-receptor for HIV. 40 Our work, as well as work with our collaborators on a related monocyte-tropic pathogen (Ehrlichia muris), demonstrates that infection with an obligate intracellular bacterial pathogen can have a similar viral-like impact on hematopoiesis.22,41 Anaplasma phagocytophylum and viral pathogens both have an intracellular life cycle and a trend towards disturbing hematopoiesis, resulting in bone marrow dysfunction and cytopenias, rather than stimulating effective hematopoiesis. Although acute A. phagocytophilum infection clearly results in HSPC proliferation and mobilization, other effects on hematopoiesis, including the induction of myelosuppressive chemokines, decreased bone marrow CFU activity, and a clinical presentation of bi- or pancytopenia, are more indicative of bone marrow dysfunction. 42
Acute infection with A. phagocytophilum triggers alterations in proliferation and trafficking of HSPCs. Specifically, acute infection triggers increased bone marrow HSPC synthetic activity that is first detected at day 2 post-infection and peaks at days 6–8. This burst of proliferative activity correlates with increased numbers of HSPCs in bone marrow, as well as in spleen and peripheral blood. Our previous work found substantial increases in splenic CFU activity during acute A. phagocytophilum infection, corresponding with increased splenic HSPC numbers noted in this study. 22 Both mobilization from bone marrow and proliferation within the spleen appear to play a role in expansion of splenic HSPC populations. Interestingly, CFU activity was decreased in bone marrow throughout infection in contrast to increased bone marrow HSPC numbers. Several potential factors may contribute to this disparity. Colony-forming potential does not necessarily correlate with in vivo proliferation, as demonstrated via IFN-γ effects on bone marrow; colony formation was suppressed in the face of expansion of lineage-negative, cKit-positive, Sca-1-positive populations. 43 Additionally, the earlier findings occurred during infection via tick-borne injection versus intraperitoneal inoculation, raising the possibility of differing immune responses affecting hematopoiesis, as tick saliva is immunomodulatory. 44
The relationship between downregulated CXCL12 signaling, HSPC proliferation, HSPC mobilization and the development of cytopenias during A. phagocytophilum infection is unclear. Cytopenias are detected as early as 2–3 d post-infection. 17 HSPC-related changes in the bone marrow are therefore not a likely immediate cause of acute cytopenias, but altered trafficking of mature blood cells due to downregulated CXCL12 signaling remains a potential cause. Our earlier studies found substantially increased numbers of mature neutrophils in the spleen during acute A. phagocytophilum infection; increased splenic leukocyte homing and sequestration as a result of shifts in CXCL12 gradients may, therefore, play a role in the development of leukopenia. 22 Work with a related pathogen, E. muris, has demonstrated that the functional alterations in bone marrow are accompanied by a decrease in classically defined megakaryocytic-erythroid progenitors and common myeloid progenitors, with an increase in functionally and phenotypically IFN-γ ‘activated’ (Sca-1 positive) cells that correlate with decreased bone marrow CFUs. 45 Cytopenias may also be caused by delayed hematopoietic suppression during A. phagocytophilum infection. Infection with similar tick-borne agents, particularly Ehrlichia canis, can result in chronic severe myelosuppression leading to sustained pancytopenia, and prolonged disruption of the stem/progenitor cell compartment is a plausible mechanism.46,47 Other infections with both LPS-positive and LPS-negative pathogens disrupt HSPC proliferation and mobilization kinetics, but generally the net effect is to augment innate immune cell production during infection.48–51 A causal link between the hematopoietic disturbances reported here and development of cytopenias during infection with A. phagocytophilum and similar pathogens remains to be shown.
HSPC proliferation and mobilization in response to Gram-positive, 50 Gram-negative,51,52 fungal49,53 and protist 48 pathogens has been demonstrated in vitro and in models of systemic infection. However, how these pathogens specifically impact hematopoietic signaling has not been established. In this article, we specifically implicate regulation of CXCL12/CXCR4 signaling as one mechanism by which this LPS-negative pathogen impacts hematopoiesis and hemic cell mobilization. Upstream effectors of signaling disruption remain to be elucidated. Further investigation of the mechanisms involved in these and other infection-induced hematopoietic disturbances may lead to improved therapeutic responses in the clinical setting.
Footnotes
Acknowledgements
This investigation was supported by the National Institutes of Health, Ruth L. Kirschstein National Research Service Award T32 RR207038 from the National Center for Research Resources (JLJ). We thank Carol Oxford at the University of California Davis Optical Biology Core Facility for flow cytometric sorting of HSPCs, and Michael Lamé and Naomi Walker at the University of California Davis School of Veterinary Medicine for technical assistance.
Conflicts of interest
None declared.