
Abstract
We have recently established a new experimental murine model for lipopolysaccharide (LPS)-mediated lethal shock with lung-specific injury. Severe lung injury is induced by administration of LPS into α-galactosylceramide (α-GalCer)-sensitized mice; the mice died with acute lung injury and respiratory distress within 24 h. α-GalCer activates natural killer T (NKT) cells in the lungs and liver, and induces the production of interferon (IFN)-γ. However, IFN-γ signaling is only triggered in the lungs and makes them susceptible to LPS. On the other hand, IFN-γ signaling is inhibited in liver and results in few hepatic lesions. Unlike liver NKT cells, lung NKT cells fail to produce interleukin (IL)-4, which down-regulates the IFN-γ signaling, in response to α-GalCer. The differential cytokine profile between lung and liver NKT cells may lead to organ-specific lung lesions. The experimental system using α-GalCer sensitization could be a useful experimental model for clinical endotoxic or septic shock as it presents respiratory failure, a typical manifestation in severe septic patients. In this review, key evidence and the introducuction of the detailed mechanism of LPS-mediated lung-specific injury in α-GalCer-sensitized mice is provided. In particular, the molecular background of organ-specific development of lung injury in the model is focused on.
Keywords
Introduction
Despite significant advances in intensive care unit technology and mechanical ventilatory support, mortality as a result of adult respiratory distress syndrome (ARDS) or multiorgan failure (MOF) has not changed significantly within the past two decades. The key to improving survival requires understanding of the factors that may initiate (or modulate) the injury to the lung and microvasculature in ARDS and MOF. Good animal models substantially facilitate research and provide powerful tools to resolve problems related to ARDS and MOF. 1 Furthermore, these models will be invaluable in improving our understanding of pathophysiology and in developing new and more effective therapeutic approaches. 1
Lipopolysaccharide (LPS) is known to cause systemic inflammatory response syndrome (SIRS), endotoxic shock, disseminated intravascular coagulation (DIC) and MOF. LPS-mediated lethality has been characterized by a number of laboratory models using sensitization with D-galactosamine, 2 Propionibacterium parvum, 3 Bacillus Calmette-Guérin (BCG) 4 and CpG DNA. 5 Generalized Shwartzman reaction is a potentially lethal shock reaction which can be induced by administration of LPS into LPS-primed animals at 24 h intervals. None of the above models present respiratory failure, a typical manifestation in severe septic patients. Therefore, there is a need for an animal model of severe sepsis characterized by respiratory failure bad enough to require mechanical ventilation.
α-Galactosylceramide (α-GalCer) was isolated from marine sponge Agelas mauritianus and is a specific ligand for human and mouse natural killer T (NKT) cells. It exhibits antitumor effects against several tumors in in vivo animal model with no cytotoxicity. We have recently reported that pretreatment with α-GalCer causes systemic lethal shock with severe lung injury after LPS challenge in mice and characterized the detailed mechanism of development of LPS-mediated lung injury. 6 – 8 In this review, the detailed mechanism of LPS-mediated lung injury in α-GalCer-sensitized mice is elucidated and the clinical usefulness of the model using α-GalCer sensitization in ARDS and septic shock-related research is discussed.
Establishment of a new lipopolysaccharide (LPS)-mediated lethal shock model with α-galactosylceramide (α-GalCer) sensitization
Mice were sensitized by an intravenous (i.v.) injection of α-GalCer (1 µg) 12 h before LPS injection. All α-GalCer-sensitized C57BL/6 mice died within 24 h after an i.v. injection of 1 µg of LPS, whereas α-GalCer-sensitized BALB/c mice required 5 µg of LPS as the lethal dose. C57BL/6 mice seemed more susceptible to LPS-mediated lethal shock than BALB/c mice. Simultaneous administration of α-GalCer and LPS was ineffective for the development of lethal shock. Successful sensitization with α-GalCer for the development of lethal shock required an interval period from 3 to 24 h between the injections of α-GalCer and LPS. For further characterization, α-GalCer (1 µg) was administered i.v. 12 h before LPS treatment. Lung injury was severe compared with the injuries of other organs, e.g. liver. There was a marked accumulation ofanumber of infiltrates in the lungs consisting of polymorphonuclear leukocytes and mononuclear cells (Figure 1). Hemorrhage and cell death were also observed. On the other hand, there was no such histologic change in the lungs of mice receiving LPS injection alone. Hepatic lesions in those mice were characterized by focal infiltration of a small number of inflammatory cells, such as polymorphonuclear leukocytes and mononuclear cells (Figure 1). Hepatic lesions were only marginal compared with lung lesions. In other organs there were no significant histologic changes with the exception of congestion.
Lipopolysaccharide (LPS)-mediated lesions in the lungs and livers of α- galactosylceramide (GalCer)-sensitized mice. The lungs and livers were removed 8 h after LPS injection. Lung, x 100 and 400; Liver, x 200. Note that the region with hepatic lesions is shown, although hepatic lesions are hardly detected.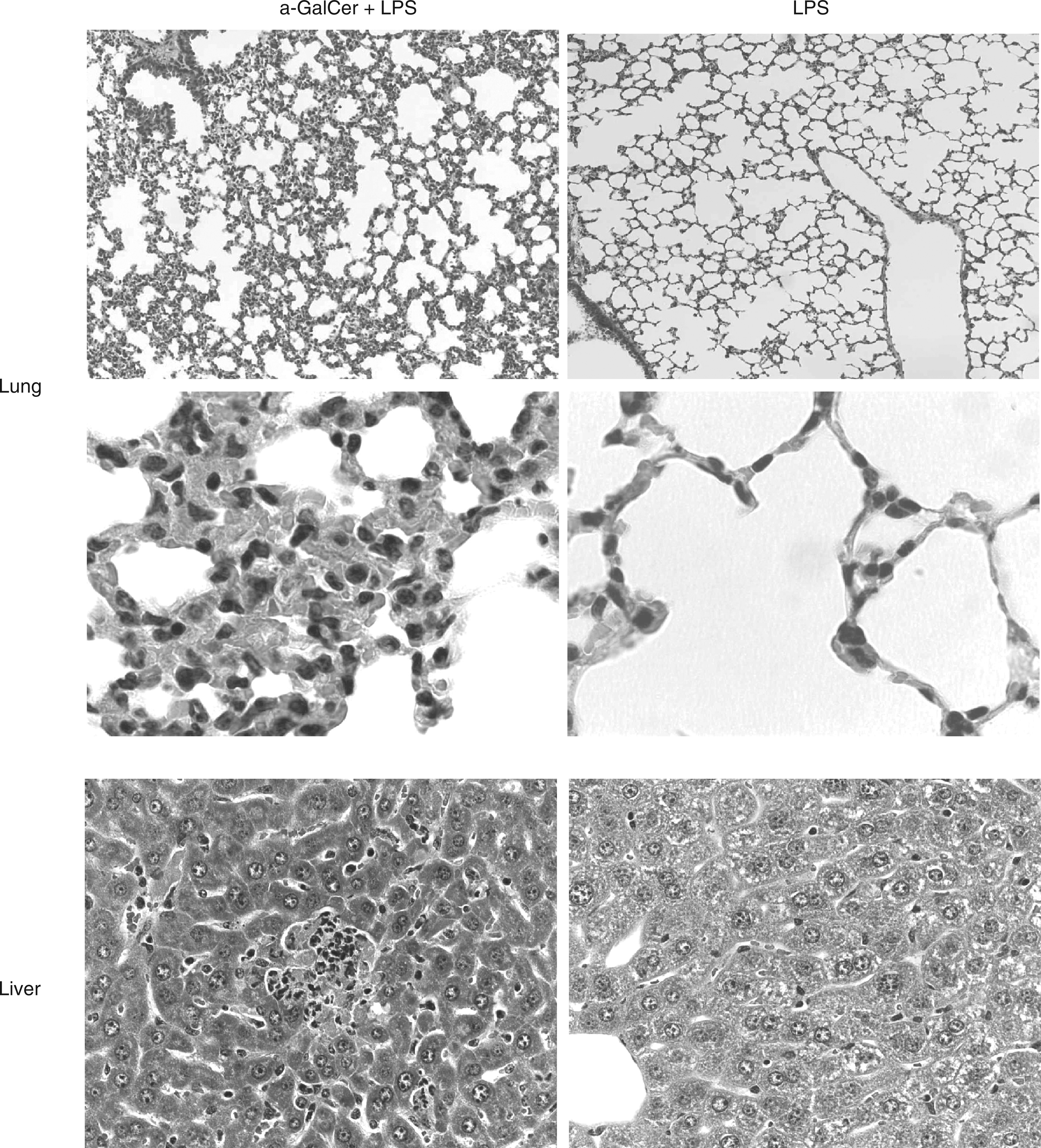
Involvement of Vα14-positive natural killer T (NKT) cells
The LPS injection killed none of the Vα14-positive NKT-deficient mice sensitized with α-GalCer, and no significant histologic change was seen in the lungs of the mice. On the other hand, LPS killed all wild-type BALB/c mice sensitized with α-GalCer. It is consistent with the fact that α-GalCer activates Vα14-positive NKT cells. 9 – 11 Administration of a high dose (1 mg) of LPS killed both Vα14-positive NKT-deficient and wild-type mice without α-GalCer sensitization, suggesting no difference in susceptibility to LPS.
Respective roles of interferon (IFN)-γ and tumor necrosis factor (TNF)-α in lipopolysaccharide (LPS)-induced lung injury
A high level of circulating interferon (IFN)-γ was detected in α-GalCer-sensitized mice before LPS treatment, suggesting that α-GalCer sensitization alone leads to a high output of circulating IFN-γ. On the other hand, the injection of LPS alone induced only a marginal amount of IFN-γ and did not affect the level of circulating IFN-γ which was induced by α-GalCer. In addition, α-GalCer did not lead to IFN-γ production in Vα14-positive NKT-deficient mice. Tumor necrosis factor (TNF)-α was not detected in α-GalCer-sensitized mice before LPS treatment, suggesting that α-GalCer induced no TNF-α production. LPS markedly augmented TNF-α production in α-GalCer-sensitized mice 1 h after administration and the level of TNF-α rapidly decreased within 3 h. However, LPS led to a lower amount of TNF-α in α-GalCer-untreated mice.
α-Galactosylceramide is known to activate NKT cells and cause release of interferon (INF)-γ. 9 – 11 In order to clarify the involvement of IFN-γ in the development of lethal shock, the preventive effect of anti-IFN-γ antibody or anti-TNF-α antibody on lethal shock was examined. Anti-IFN-γ antibody completely prevented α-GalCer-sensitized mice from LPS-mediated lethal shock and the development of lung lesions was completely abolished. Anti-TNF-α antibody also rescued four out of six mice from LPS-mediated lethal shock. Thus, the inhibitory effect of anti-TNF-α antibody was partial; however, TNF-α is known as a factor in LPS-mediated lethality.12, 13 Administration of LPS did not kill α-GalCer-sensitized TNF-α-deficient B10D2 mice but it did, however, kill α-GalCer-sensitized wild-type B10D2 mice. Based on the finding that TNF-α-deficient mice produced IFN-γ normally in response to α-GalCer, the experimental result clearly confirmed the involvement of TNF-α in lethal shock. α-GalCer-induced IFN-γ production and LPS-induced TNF-α production are suggested to play a pivotal role in the sensitization and induction of lung injury, respectively.
Development of lipopolysaccharide (LPS)-mediated lung injury in α-galactosylceramide (α-GalCer)-sensitized mice
Role of interferon (IFN)-γ in the sensitization of lethal shock
The number of NKT cells expressing pan-NK markers and CD3 increased in the lungs of α-GalCer-sensitized mice. There were a small number of such cells in the lungs of normal control mice. The increase of NKT cells expressing both NK and T cell markers was reasonable as α-GalCer is the ligand stimulating NKT cells. Moreover, NKT cells present in the lungs of α-GalCer-sensitized mice were larger in size than the cells from normal control mice, suggesting the presence of activated NKT cells in the lungs of α-GalCer-sensitized mice. 7 A high level of IFN-γ mRNA expression was detected in the lungs 3 h after the injection of α-GalCer and as such, IFN-γ mRNA expression continued up to 12 h. Only slight IFN-γ mRNA expression was detected 6 h after injection with LPS alone.Intracellular IFN-γ-producing cells increased in the lungs of α-GalCer-pretreated mice. Therefore, α-GalCer is suggested to induce local IFN-γ production via the activation of NKT cells present in the lungs.
Interestingly, a significant number of inflammatory cells were recruited into the lungs of α-GalCer-sensitized mice. The cell numbers in bronchoalveolar lavage fluid (BALF) from α-GalCer-treated and untreated mice were approximately 51,000 ± 1,000/lung and 21,000 ± 500/lung, respectively, and approximately 85% of the infiltrates were mononuclear cells. 7 Vascular cell adhesion molecule (VCAM)-1 mRNA expression was strikingly up-regulated in the lungs of α-GalCer-sensitized mice. The expression of E-selectin mRNA was also augmented. On the other hand, ICAM-1 mRNA expression did not significantly increase in α-GalCer sensitization. The up-regulation of VCAM-1 mRNA expression was definitely inhibited by anti-IFN-γ neutralizing antibody. High expression of VCAM-1 was detected in blood vessels in the lungs 6 h after α-GalCer sensitization and continued up to 12 h. The expression of ICAM-1 and E-selectin was detected as a faint positive staining. Anti-VCAM-1 antibody significantly prevented the mice from LPS-induced lung injury and lethality (three out of six mice), suggesting that VCAM-1 has a pivotal role.
Expression of VCAM-1 is known to recruit cells expressing very late antigen (VLA)-4 as the counterpart.14, 15 The level of VLA-4 mRNA expression was markedly augmented in the lungs 9 h after α-GalCer treatment, and further increased up to 12 h. LPS alone did not affect VLA-4 mRNA expression. VLA-4 was definitely expressed on infiltrates in the lungs of α-GalCer-sensitized mice.
Downstream pathologic effects of lipopolysaccharide (LPS)-mediated lethal shock after α-galactosylceramide (α-GalCer)sensitization
Injection of LPS led to a higher production level of TNF-α in lung tissues and BALF from α-GalCer-sensitized mice than in untreated mice. A higher level of IL-1α was also detected in the sera, lung tissue and BALF of α-GalCer-sensitized mice. LPS induced a higher output of IL-6 as well as TNF-α and IL-1α in α-GalCer-sensitized mice. In addition, the expression of TNF-α and IL-6 mRNA was up-regulated in the lungs after the administration of LPS into α-GalCer-sensitized mice.
There are several reports supporting the participation of nitric oxide (NO) and its rapid reaction with superoxide, leading to generation of extremely harmful oxidants, such as peroxynitrite and hydroxyl radical, in sepsis-induced acute lung injury.16, 17 The administration of LPS induced a marked expression of an inducible type of NO synthase (iNOS) mRNA in α-GalCer-sensitized mice. In addition, the injection of α-GalCer alone did not induce it in mice. The NO-mediated tissue damage could be confirmed with anti-nitrotyrosine antibody. LPS caused severe lung injury in α-GalCer-sensitized mice via the production of proinflammatory cytokines and NO. Administration of LPS led to the leakage of proteins and inflammatory cells into the BALF in the lungs of α-GalCer-sensitized mice, suggesting elevated pulmonary permeability.
The mechanism of development of lung-specific injury
α-GalCer has been identified as a glycolipid ligand, which stimulates a special group of NKT cells. 9 – 11 NKT cells rapidly produce IFN-γ, IL-4 and IL-10 in response to α-GalCer. 18 The differential response in the development of lesions between the lung and the liver to α-GalCer may be dependent on the functional difference in α-GalCer-stimulated NKT cells between the two organs.
α-Galactosylceramide (α-GalCer) induces interferon (IFN)-γ production in both lung and liver natural killer T (NKT) cells
NKT cells are known to produce IFN-γ in response to α-GalCer. 11 Intracellular IFN-γ was markedly expressed in both NKT cells from the lungs and liver of α-GalCer-treated mice. There was no significant difference in intracellular IFN-γ expression between lung and liver NKT cells. α-GalCer significantly induced the expression of IFN-γ protein and mRNA in the lungs and liver.
Interferon (IFN)-γ signaling is triggered in the lungs but not liver of α-galactosylceramide (α-GalCer)-sensitized mice
Although IFN-γ was produced in both the lungs and liver of α-GalCer-injected mice, successive IFN-γ signaling was only triggered in the lungs. The expression of phosphorylated signal transducer and activator of transcription (STAT)1 was significantly augmented in the lungs and liver 4 h after α-GalCer injection. Augmented expression of phosphorylated STAT1 in lungs continued up to 12 h, whereas expression in liver clearly declined at 8 h. Interferon regulatory factor (IRF)1 expression was significantly augmented in the lungs but not in liver. Successive IFN-γ signaling was characteristic in the lungs of α-GalCer-injected mice.
Liver natural killer T (NKT) cells but not lung NKT cells produce interleukin (IL)-10 in response to α-galactosylceramide (α-GalCer)
INF-γ signaling was triggered in the lungs but not the liver of α-GalCer-injected mice. Therefore, the possibility that IFN-γ signaling in liver may be inhibited by an anti-inflammatory cytokine, such as IL-10, was raised. IFN-γ production and signaling occurred 4 h after α-GalCer stimulation. Cytokines affecting IFN-γ production and signaling must be produced within 4 h, therefore, further characterization on the action of IL-10 and IL-4 was examined 2 h after α-GalCer stimulation. Liver NKT cells expressed intracellular IL-10, whereas lung NKT cells did not. IL-10 mRNA was clearly expressed in liver NKT cells but not in lung NKT cells. Impaired IFN-γ signaling in the liver of α-GalCer-sensitized mice may be responsible for IL-10 production by liver NKT cells.
Interleukin (IL)-10 is constitutively expressed in normal lungs
Phosphorylation of STAT3 was clearly detected in the liver of α-GalCer-sensitized mice at 4 h, after which it gradually declined. Suppressor of cytokine signaling (SOCS)3 expression was significantly augmented 8 h after α-GalCer injection and further increased up to 12 h. Surprisingly, SOCS3 expression and STAT3 phosphorylation were also detected in the lungs even before α-GalCer injection, indicating a constitutive IL-10 expression in lungs. Phosphorylation of STAT3 was augmented 4 h after α-GalCer treatment and gradually decreased until 12 h. Lung alveolar epithelial cells were positively stained with anti-IL-10 antibody. There was no significant difference in IL-10 expression in the lung extracts between α-GalCer-treated and untreated mice. There was no positive IL-10 staining in normal liver sections. Thus, IL-10 was constitutively expressed on alveolar epithelial cells in normal lungs.
Anti-IL-10 neutralizing antibody significantly enhanced IFN-γ production in both lungs and liver of α-GalCer sensitized mice, suggesting that IL-10 inhibited IFN-γ production in those mice. However, the differential response in IFN-γ signaling between the lungs and liver could not be elucidated by the production of anti-inflammatory IL-10.
Natural killer T (NKT) cells in liver, but not the lungs, produce interleukin (IL)-4 in response to α-galactosylceramide (α-GalCer)
IL-4 inhibits IFN-γ signaling 19 and NKT cells produce IL-4 in response to α-GalCer. 20 Therefore, IL-4 MAY be involved in the differential response of lungs and liver to IFN-γ. In fact, liver NKT cells in α-GalCer-sensitized mice significantly expressed intracellular IL-4, whereas lung NKT cells did not. IL-4 mRNA was highly expressed in liver NKT cells but not in lung NKT cells from α-GalCer-sensitized mice.
SOCS1 expression was clearly detected in liver homogenates 4–12 h after α-GalCer injection. On the other hand, only slight SOCS1 expression was detected in lung homogenates at 8 and 12 h. Expression of phosphorylated STAT6 was much more augmented in liver homogenates 4–12 h after α-GalCer injection than in lung homogenates. IL-4 production in liver NKT cells was suggested to inhibit IFN-γ signaling via augmented SOCS1 and phosphorylated STAT6 expression. On the other hand, no IL-4 production in lung NKT cells may allow the triggering of IFN-γ signaling in the lungs. Thus, IL-4 may be a responsible effector molecule for the differential response of liver and lung to IFN-γ.
Neutralization of interleukin (IL)-4 causes severe liver injury in α-galactosylceramide (α-GalCer)-sensitized mice
Administration of LPS into α-GalCer-sensitized mice caused the appearance of a number of apoptotic cells in the lungs but not in liver. Anti-IL-4 neutralizing antibody did not affect the number of apoptotic cells in the lungs, whereas it caused the appearance of a number of apoptotic cells in liver. Thus, it was suggested that IL-4 plays a crucial role in preventing liver injury in α-GalCer-sensitized mice. Anti-IL-4 neutralizing antibody augmented the phosphorylation of STAT1 and reduced the expression of SOCS1 in liver. On the other hand, neither anti-IL-4 antibody nor anti-IL-10 antibody affected them in the lungs. Therefore, it was suggested that IL-4 prevented hepatic lesions in α-GalCer-sensitized mice through the inhibition of IFN-γ signaling.
CD8+ natural killer T (NKT) cells are present in the lungs but not liver of normal mice
The differential cytokine profile between lung and liver NKT cells suggested a difference in their phenotypes. A significant number of CD8+ cells were detected in lung NKT cells, whereas few such cells were detected in liver; however, liver NKT cells mainly consisted of CD4+ cells.
Discussion
Liver NKT cells produce IL-4 in response to α-GalCer and IL-4 prevents hepatic lesions through the inhibition of IFN-γ signaling. Lung NKT cells fail to produce IL-4 in response to α-GalCer, which inhibits the IFN-γ signaling Therefore, IFN-γ signaling is exclusively triggered in the lungs of α-GalCer-sensitized mice and results in the expression of adhesion molecules, such as VCAM-1, on vascular endothelial cells in the lungs.
7
Subsequently, VCAM-1 expression recruits inflammatory cells expressing VLA-4 as the counterpart of VCAM-1 in the lungs and those cells produce an excess of TNF-α in response to LPS.
7
The release of excessive TNF-α leads to the elevation of pulmonary permeability and massive cell death, followed by severe lung-specific injury.
7
This is a putative mechanism of the development of lung lesions in LPS-mediated lethal shock using α-GalCer sensitization (Figure 2).
A putative mechanism of lipopolysaccharide (LPS)-mediated lung-specific injury in α- galactosylceramide (GalCer)-sensitized mice. IFN, interferon; IL, interleukin; NKT, natural killer T cell; SOCS1, suppressor of cytokine signaling 1; VCAM-7, Vascular cell adhesion molecule 7; VLA-4, very late antigen.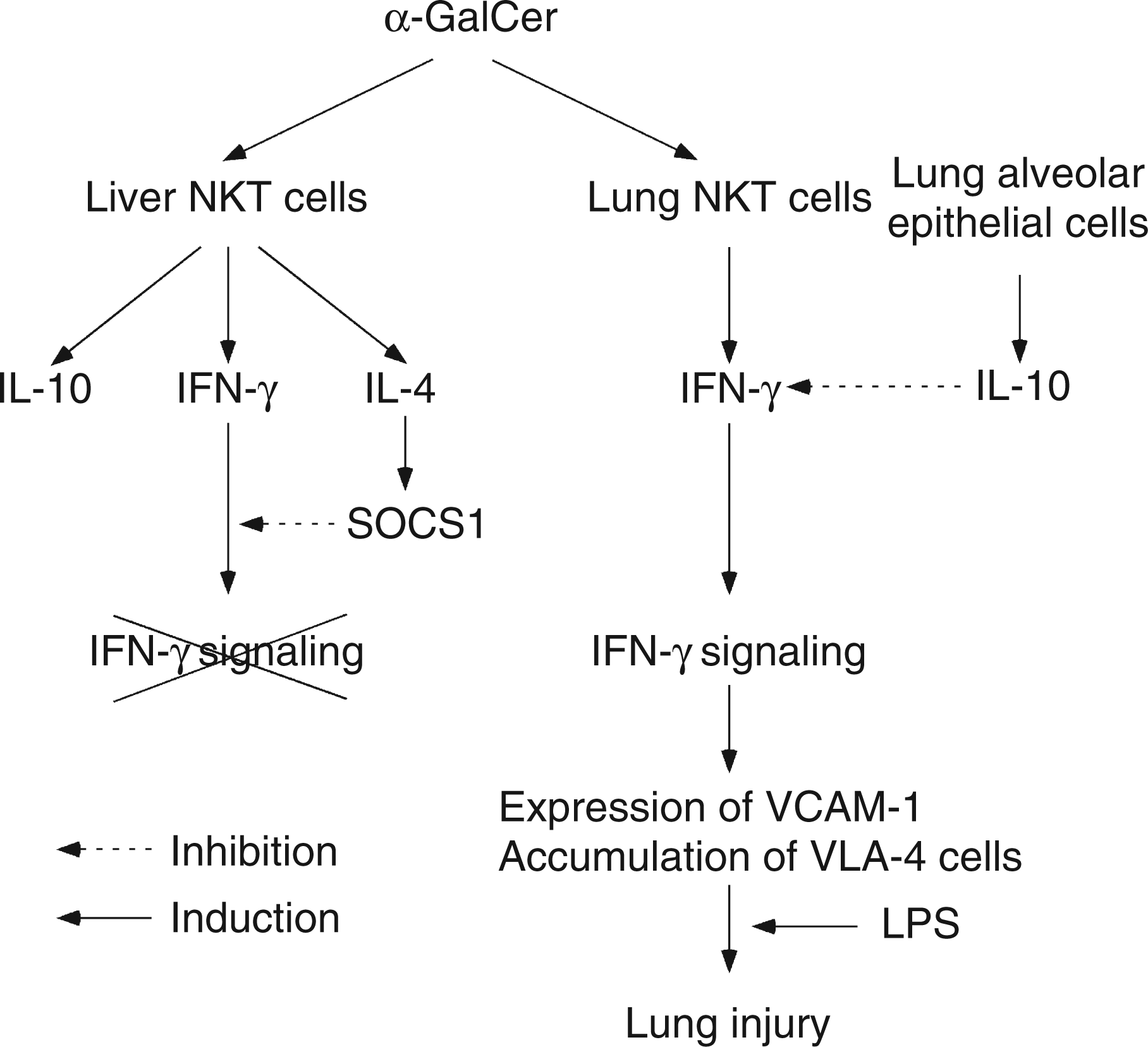
IL-4 prevents the development of hepatic lesions via inhibition of IFN-γ signaling in α-GalCer-sensitized mice. IL-4 is reported to inhibit IFN-γ signaling through the activation of STAT6. 19 Furthermore, IL-4 prevents IFN-γ signaling via augmented expression of SOCS1, which negatively regulates IFN-γ signaling. SOCS1 is known to inhibit IFN-γ signaling by directly binding to the IFN-γ receptor. 21 – 23 Moreover, mice lacking a functional SOCS1 gene are hypersensitive to IFN-γ and have a number of characteristic lesions, such as hepatic necrosis, macrophage infiltration in several organs, multiple hematopoietic abnormalities and severe lymphopenia.24, 25 Thus, IL-4 produced by liver NKT cells is suggested to inhibit IFN-γ signaling via STAT1 inactivation and SOCS1 induction. It is also supported by the finding that anti-IL-4 antibody enhances STAT1 activation in IFN-γ signaling and exacerbates hepatic lesions. On the other hand, the failure of IL-4 production in lung NKT cells in response to α-GalCer is unable to inhibit IFN-γ signaling and causes severe lung lesions. Thus, IL-4 may be a main regulatory molecule in the development of LPS-mediated lung-specific lesions in α-GalCer-sensitized mice.
Lung NKT cells exclusively produce IFN-γ, whereas liver NKT cells produce IL-4 and IL-10, as well as IFN-γ. Considering that IL-4 is the crucial molecule for the inhibition of LPS-induced hepatic lesions in α-GalCer-sensitized mice, the differential cytokine profile between lung and liver NKT cells may be responsible for the development of lung-specific injury. In fact, it has been reported that NKT cells are phenotypically, functionally and developmentally heterogeneous, and that three distinct subsets consisting of double negative CD4+ and CD8+ are differentially distributed in a tissue-specific fashion.26–28 The majority of NKT cells in most tissues are either CD4+ or double negative. 29 However, CD4+ and CD8+ NKT cells are present in all tissues except the thymus in mice,27,28,30, 31 and are especially dominant in thelungs. 27 CD4+ or double negative NKT cells produce IL-4 as well as IFN-γ, whereas CD8+ NKT cells only produce IFN-γ. 30 – 32 It supports the possibility that lung NKT cells may express CD8+ and exclusively produce IFN-γ in response to α-GalCer. The differential cytokine profile between lung and liver NKT cells is suggested to cause the differential response between the lungs and liver to IFN-γ and exclusively develop lung lesions in α-GalCer-sensitized mice.
As far as is known, there is no experimental model of LPS-mediated lethal shock with lung-specific injury. Previously reported LPS-mediated lethal shock models are characterized by hepatic injury 2 – 5 . Sensitization of mice with D-galactosamine as a hepatotoxic agent augments the susceptibility of hepatocytes to TNF-α toxicity. 2 Sensitization with P. parvum, 3 BCG 4 or CpG DNA 5 may activate macrophages in the liver via hepatic uptake; those activated macrophages may produce a locally high level of TNF-α in response to LPS, followed by hepatic injury. On the other hand, effector cells in α-GalCer sensitization are NKT cells. The characteristic cytokine production of lung NKT cells may incidentally cause novel organ-specific lung injury.
In a clinical setting, patients with severe septic shock are characterized by respiratory dysfunction, accompanied by lung lesions. The activation of lung NKT cells in pulmonary infections may lead to the production of IFN-γ without IL-4 capable of inhibiting IFN-γ signaling and the development of lung lesions. Moreover, more severe lung lesions would develop in patients if constitutive IL-10 synthesis was impaired in alveolar epithelial cells. A series of complicated mechanisms may regulate the development of severe lung lesions in clinical septic shock. The involvement of NKT cells in the development of lung injury in human septic shock requires further characterization.
Conclusion
The LPS-mediated lethal shock model using α-GalCer sensitization is characterized by severe lung-specific injury. Lung NKT cells play a critical role in the development of lung injury via IFN-γ production. IFN-γ signaling is triggered in the lungs as lung NKT cells do not produce IL-4, which down-regulates IFN-γ signaling. IFN-γ signaling leads to severe lung injury via a series of inflammatory responses. The experimental murine model with severe lung-specific injury may provide a powerful tool for the analysis of clinical endotoxic or septic shock.
Footnotes
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan. I am grateful to Jargalsaikhan Dagvadorj, Gantsetseg Tumurkhuu, Hiroyasu Ito and Naoki Koide for their assistance in the department.