
Abstract
During acute inflammation, monocytes are essential in abolishing invading micro-organisms and encouraging wound healing. Recruitment by CC chemokines is an important step in targeting monocytes to the inflamed tissue. However, cell surface expression of the corresponding chemokine receptors is subject to regulation by various endogenous stimuli which so far have not been comprehensively identified. We report that the platelet-derived CXC chemokine ligand 4 (CXCL4), a known activator of human monocytes, induces down-regulation of CC chemokine receptors (CCR) 1, −2, and −5, resulting in drastic impairment of monocyte chemotactic migration towards cognate CC chemokine ligands (CCL) for these receptors. Interestingly, CXCL4-mediated down-regulation of CCR1, CCR2 and CCR5 was strongly dependent on the chemokine’s ability to stimulate autocrine/paracrine release of TNF-α. In turn, TNF-α induced the secretion CCL3 and CCL4, two chemokines selective for CCR1 and CCR5, while the secretion of CCR2-ligand CCL2 was TNF-α-independent. Culture supernatants of CXCL4-stimulated monocytes as well as chemokine-enriched preparations thereof reproduced CXCL4-induced CCR down-regulation. In conclusion, CXCL4 may act as a selective regulator of monocyte migration by stimulating the release of autocrine, receptor-desensitizing chemokine ligands. Our results stress a co-ordinating role for CXCL4 in the cross-talk between platelets and monocytes during early inflammation.
Introduction
In addition to their roles in haemostasis, thrombosis and tissue repair, platelets have important functions in initiating an inflammatory response. 1 Activated human platelets release a variety of mediators that have the capacity to induce the primary steps of inflammation, e.g. to recruit blood leukocytes to sites of vascular tissue damage.2,3 Prominent platelet-secreted mediators involved are several chemokines, such as CXC chemokine ligand 7 (CXCL7), which have been shown to attract neutrophil granulocytes (neutrophils) selectively, while others such as CCL3 and CCL5 are potent monocyte attracting chemokines. 4 – 6 A very complex role has been assigned to CXCL4, also referred to as platelet factor 4 (PF-4), which is a very unusual chemokine in that it lacks chemo-attractant activity for leukocytes, including neutrophils, monocytes, and T-cells.3,4,7– 9 Instead, CXCL4 participates in the control of acute cellular defence mechanisms as well as in long-term regulation of various immune and other cells. This is supported by findings describing CXCL4-mediated induction of histamine release in basophils, activation of secondary granule exocytosis and cell adhesion to endothelium in neutrophils, suppression of IL-2 production in human T-cells and concomitant inhibition of T-cell proliferation, as well as control of endothelial cell (EC) and fibroblast proliferation.7,9– 13 An especially sophisticated array of consecutive functions is subject to CXCL4 control in monocytes. These cells, that act as first-line mediators in inflammation and at the same time are major players in mounting an adaptive immune response, react to CXCL4 not only by short-term antimicrobial responses such as oxygen radical formation and phagocytosis or by rapid production of TNF-α, but thereafter undergo a cellular programme resulting in prevention of spontaneous apoptosis and in the differentiation of these cells into a specific subtype of macrophages with enhanced innate immune function.8,14 Interestingly, we found monocyte responses to CXCL4 to be independent of Gi-proteins or elevation of intracellular free calcium, which is surprising since chemokines typically act on cells through binding to 7-transmembrane-domain G protein-coupled receptors (GPCR). 8 In fact, binding sites for CXCL4 are less well defined. While in a recent report Lasagni et al. 15 described an alternatively spliced variant of CXCR3, also referred to as CXCR3-B, as a functional receptor for CXCL4 on endothelial cells; our own investigations revealed that CXCR3-B is not expressed on monocytes or neutrophils, and that CXCL4-binding to the latter cells is mediated by a distinct receptor, recently identified as a chondroitin sulphate proteoglycan.8,14,16, 17 While interaction of CXCL4 with such types of receptor may be responsible for its inability to act as an chemo-attractant for leukocytes, the chemokine nevertheless has a regulatory impact on cell migration through modulating upstream events. Nesmelova et al. 18 described enhancement of CXCL8-dependent chemotaxis following heterodimerization of the chemokine with CXCL4. According to our own results, CXCL4 promotes adhesion of neutrophils to unstimulated endothelium and at substimulatory concentrations drastically enhances CXCL7-induced adhesion. 19 Furthermore, CXCL4 enhances the arrest of CCL5-stimulated monocytes on activated endothelium under flow conditions, which is likely also related to direct molecular interaction of the chemokines. 20 However, a more common target in the regulation of cell migration are chemokine receptors themselves, especially the regulation of their cell surface expression and functionality. Specifically for monocytes, it is known that migration to distinct tissues/organs, determining their functional maturation, crucially depends on chemokine receptor expression.21,22 Regarding the importance of CXCL4 as a monocyte-directed mediator, we therefore were interested in its ability to modulate chemokine receptor expression on these cells and to concomitantly alter their chemotactic response.
As the aim of our present study was to elucidate further CXCL4’s role during an acute inflammatory situation, we specifically investigated its impact on receptors CCR1, CCR2, and CCR5, which all bind CC-chemokines involved in the recruitment of monocytes to inflammatory sites. 23 As our results demonstrate, CXCL4 effectively down-modulates cell surface expression of all three receptors on monocytes, resulting in reduced chemotactic activity of the cells towards receptor-specific ligands CCL2 (CCR2), CCL3 and CCL5 (CCR1, CCR-5). Interestingly, CXCL4 appears to down-modulate receptor expression via indirect mechanisms, involving the autocrine induction and secretion of CCR ligands CCL2, CCL3, and CCL4 as well as of TNF-α in monocytes, while CCL5 is not involved. Thus a role for CXCL4 in vascular inflammation may be to retain recruited monocytes at sites of vascular inflammation and at the same time to promote a second wave of leukocyte recruitment through chemokine induction in the former cells.
Materials and methods
Cytokines
Human CXCL4 was purified to homogeneity from release supernatants of thrombin-stimulated platelets in a three-step procedure as previously described.7,16 The preparations contained <0.125 ng LPS/mg CXCL4 (i.e. below 4 pg/ml at 4 µ
Cell preparation, culture, and stimulation
Mononuclear cells (MNCs) were isolated from peripheral blood of single healthy donors by Ficoll-Paque (Pharmacia LKB, Freiburg, Germany) gradient centrifugation. Monocytes and lymphocytes were separated by counter flow centrifugation as described previously.
25
The prepared monocyte fraction consisted of more than 95% CD14 positive cells in all events as determined by flow cytometry analysis with anti-CD14-specific monoclonal antibody (mAb; clone TüK4, IgG2A, directly PE-labelled; Dako, Glostrup, Denmark). Monocyte culture was routinely performed in RPMI-1640 supplemented with 100 U/ml penicillin G, 100 µg/ml streptomycin, 2 mmol/l
Immunofluorescence labelling and flow cytometry
Unconjugated murine mAbs directed against the following human antigens were used for indirect immunofluorescence labelling of human monocytes: CCR1 (clone 53504.111, IgG2B), CCR2 (clone 48607.121, IgG2B), CCR5 (clone 45531.111, IgG2B), CXCR4 (clone 44716.111, IgG2B), all from R&D Systems, and IgG2B control antibody (clone DK-G09; Dako, Glostrup, Denmark). Against human CCR3 a rat unconjugated mAb (clone 61828.111, IgG2A) and rat IgG2A control antibody (clone 54447.1), both from R&D Systems, were used, while a unconjugated goat antiserum (Invitrogen, Karlsruhe, Germany) served to detect human CCR4 in comparison to a normal goat control-serum (kind gift from Dr J. Gerdes, Research Center Borstel, Germany).
In experiments where cells had been pre-incubated with neutralizing anti-TNF antibodies, murine mAbs directly conjugated to PE (all from R&D Systems) were used to detect CCRs (CCR1; clone 53504.111, IgG2B), (CCR2; clone 48607.121, IgG2B), (CCR5; clone 45531.111, IgG2B), (IgG 2B control antibody; clone 20116), to circumvent the use of secondary antibodies and their potential cross-reactivity with the neutralizing antibodies. For direct immunofluorescence labelling, monocytes were incubated with the respective PE-conjugated antibodies at concentrations recommended by the manufacturer for 30 min on ice in PBS/0.1% BSA, washed with PBS/0.1% BSA, and finally resuspended in PBS/0.1% BSA. For indirect immunofluorescence labelling, cells were correspondingly incubated with the unconjugated antibodies and washed, followed by incubation with dichlorotriazinylaminofluorescein (DTAF)-conjugated goat anti-mouse, mouse anti-rat, or mouse anti-goat antibody (all from Dianova, Hamburg, Germany). After 30 min of incubation, the monocytes were washed again and finally resuspended in PBS/0.1% BSA. Flow cytometry analyses were performed on a FACSCalibur (Becton Dickinson, Heidelberg, Germany). Monocytes cultured over several days, showed a donor-dependent increase in autofluorescence. To compare analyses from different donors, data in some experiments are given as the ratio in median fluorescence intensity (rMFI), calculated by dividing the median fluorescence intensity (MFI) of cells labelled with specific antibody by the MFI of cells labelled with irrelevant (isotype) antibody.
Measurement of chemokine release by ELISA
Cell-free supernatants of monocytes stimulated with CXCL4 or TNF-α for 2–72 h were harvested and stored at −20°C until analysis. Concentrations of CCL5, CCL2, CCL3 and CCL4 were determined using specific quantitative enzyme-linked immunosorbent assays (ELISAs), all purchased from R&D Systems, according to the manufacturer’s advice. For the measurement of CXCL8, an ELISA, developed in our laboratory, was used as described. 24
Immunodepletion of monocyte culture supernatants with specific chemokine antibodies
Cell-free supernatants of monocytes stimulated with CXCL4 or left unstimulated for 10 h were harvested and divided into 5-ml aliquots. Aliquots received either a combination of neutralizing monoclonal antibodies to CCL2 (5 µg/ml of clone 24822), CCL3 (3 µg/ml of clone 93321) and CCL4 (2.5 µg/ml of clone 24006), all from R&D systems, or the same amounts of control antibodies of the corresponding isotype. After incubation for 45 min at room temperature, samples were diluted 1 : 2 with buffer (0.1
Monocyte chemotaxis
Monocyte migration in response to chemokines was assayed using 24-well Transwell plates (Costar, Bodenheim, Germany) with a polycarbonate membrane (6.5 mm diameter, pore size 5.0 µm) at the bottom of the inserts. Briefly, the lower compartment was filled with 600 µl chemokines diluted in RPMI-1640/10% FCS and equilibrated for 20 min (5% CO2, 37°C). Subsequently, inserts received 100 µl cell suspension (8 × 106 monocytes/ml RPMI-1640/10% FCS). After 2 h, Transwell plates were placed on ice, cells left in the inserts were removed, and 100 µl ice-cold PBS was added to the inserts for 45 min. After washing with ice-cold PBS, inserts received 100 µl of cell dissociation solution (non-enzymatic; Sigma, Munich, Germany) for 30 min at 37°C to detach transmigrated monocytes still adherent to the lower side of the membrane. After centrifuging Transwell plates for 10 min at 300 g, inserts were removed and cells in the lower compartment were lysed with Triton X-100 (0.1% final). According to cell numbers counted after May-Gruenwald-staining of membranes, <1% of cell input remained attached to the lower side of the membranes. Endogenous β-glucuronidase enzymatic activity was measured using p-nitrophenyl-β-glucuronide (Sigma) as a substrate as described. 19 The number of migrated cells was calculated by means of a standard of lysed cells run in parallel. In some experiments, we attempted to neutralize the chemotactic activity contained in supernatants from CXCL4-stimulated monocytes by adding 3 µg/ml of inhibitory murine mAbs to human chemokines, i.e. anti-CCL5 (IgG1; clone 21418.221), anti-CCL2 (IgG1; clone 24822.111), anti-CCL3 (IgG1; clone 93321), anti-CCL4 (IgG2B; clone 24006), anti-CXCL8 (IgG1; clone 6217), all purchased from R&D Systems. Isotype controls were mouse IgG1 (clone BO7; 26 Research Center Borstel, Germany) and mouse IgG2B (clone DK-GO9; Dako).
Quantitative RT-PCR
Primers used in quantitative RT-PCR to detect chemokine mRNA expression

Quantitative RT-PCR was performed in a LightCycler instrument (Roche, Mannheim, Germany), using 2 µl cDNA and 8 µl PCR master reaction (FastStart DNA Master SYBR Green I, Roche) for the primer combinations and PCR parameters listed. Calculation was performed using LightCycler software v.3.5.3 and the second derivative maximum algorithm.
GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Statistical analysis
Data are presented as mean ± SD for the number of experiments indicated. Statistically significant (P < 0.05) differences among treatment groups were calculated using Friedman test.
Results
CXCL4 induces CC-chemokine receptor down-regulation on human monocytes
Monocytes constitutively express several receptors for inducible CC chemokines which are produced in inflammatory conditions. Thus, to investigate a potential impact of CXCL4 on CC chemokine-mediated monocyte activation, we examined whether CXCL4 has the capacity to modulate surface expression of CC chemokine receptors on these cells. For this, freshly elutriated human monocytes were stimulated with CXCL4 or left untreated, and surface expression of CCR1, CCR2, CCR3, CCR4, and CCR5 was assessed by flow cytometry at the beginning (time point 0) and after 5 min, 30 min, 2 h and 18 h of culture. For a comparison, the expression of CXCR4, a chemokine receptor having a role in steady-state turnover of leukocytes rather than affecting their traffic to inflammatory sites, was monitored in parallel. A constant concentration of 4 µM CXCL4 was chosen for these experiments, because this dosage has been shown previously to induce optimal functional responses in human monocytes.8,14
As expected, untreated monocytes at time point 0 clearly exhibited surface expression of CCR1 (rMFI = 8.1 ± 3.2), CCR2 (rMFI = 10.5 ± 3.9), CCR5 (rMFI = 5.6 ± 1.6), and CXCR4 (rMFI = 14.5 ± 4.8), whereas CCR3 and CCR4 were not significantly expressed (Fig. 1A). The latter two receptors remained undetectable throughout the entire culture period on untreated as well as on CXCL4-stimulated monocytes, indicating that CXCL4 is unable to induce CCR3 and CCR4 surface expression. Unstimulated monocytes underwent no significant changes in CCR1, CCR2, CCR5 and CXCR4 expression during the first 2 h of culture, whereas after 18 h there was a 1.4-fold increase in CCR1 (rMFI = 11.5 ± 3.3), a 2.1-fold increase in CCR5 (rMFI = 11.6 ± 3.0), a 1.7-fold increase in CXCR4 (rMFI = 25.8 ± 12.8), and no change (1.05-fold) in CCR2 (rMFI = 11.0 ± 5.0; Fig. 1A). The CXCL4-stimulated monocytes likewise exhibited no significant changes within the first 2 h, showing the same degree of receptor expression as unstimulated monocytes, whereas after 18 h all chemokine receptors except CXCR4 had undergone drastic down-regulation, as compared to the unstimulated monocytes cultured for the same time period. Thus, surface expression of CCR1 appeared reduced by 65 ± 12%, that of CCR2 by 63 ± 14%, and that of CCR5 by 62 ± 28%, while CXCR4 remained unaffected. For better comparison, histograms of the latter results (one representative experiment) are given in Figure 1B. While these data demonstrate a negative regulatory impact of CXCL4 on CCR expression, they also show that CXCL4 did not simply prevent culture-associated receptor up-regulation, since expression of all CC chemokine receptors after 18 h was also significantly lower than at time point 0. To verify the functionality of monocyte-expressed CCR1, CCR2, CCR5, and CXCR4, cells in parallel were incubated with cognate chemokine ligands for these receptors, i.e. CCL5, CCL2, and CXCL12 (Fig. 1A). Exposure to CCL5 led to rapid and drastic down-regulation of CCR1 after 2 h and of CCR5 after 30 min already, while exposure to CCL2 and CXCL12 induced down-regulation to comparable degrees after 2 h of CCR2 and CXCR4, respectively. Interestingly, surface expression of these receptors (almost) completely recovered after 18 h of incubation to the levels seen in untreated cells after that time. Thus, apart from demonstrating the responsivity of the receptors to their ligands, these results also revealed a striking difference in the time kinetics of chemokine ligand and CXCL4-induced receptor regulation.
Impact of CXCL4 on CC chemokine receptor surface expression in monocytes. (A) Monocytes (106/ml) were cultured for up to 18 h in the absence (empty bars) or presence (solid bars) of 4 µ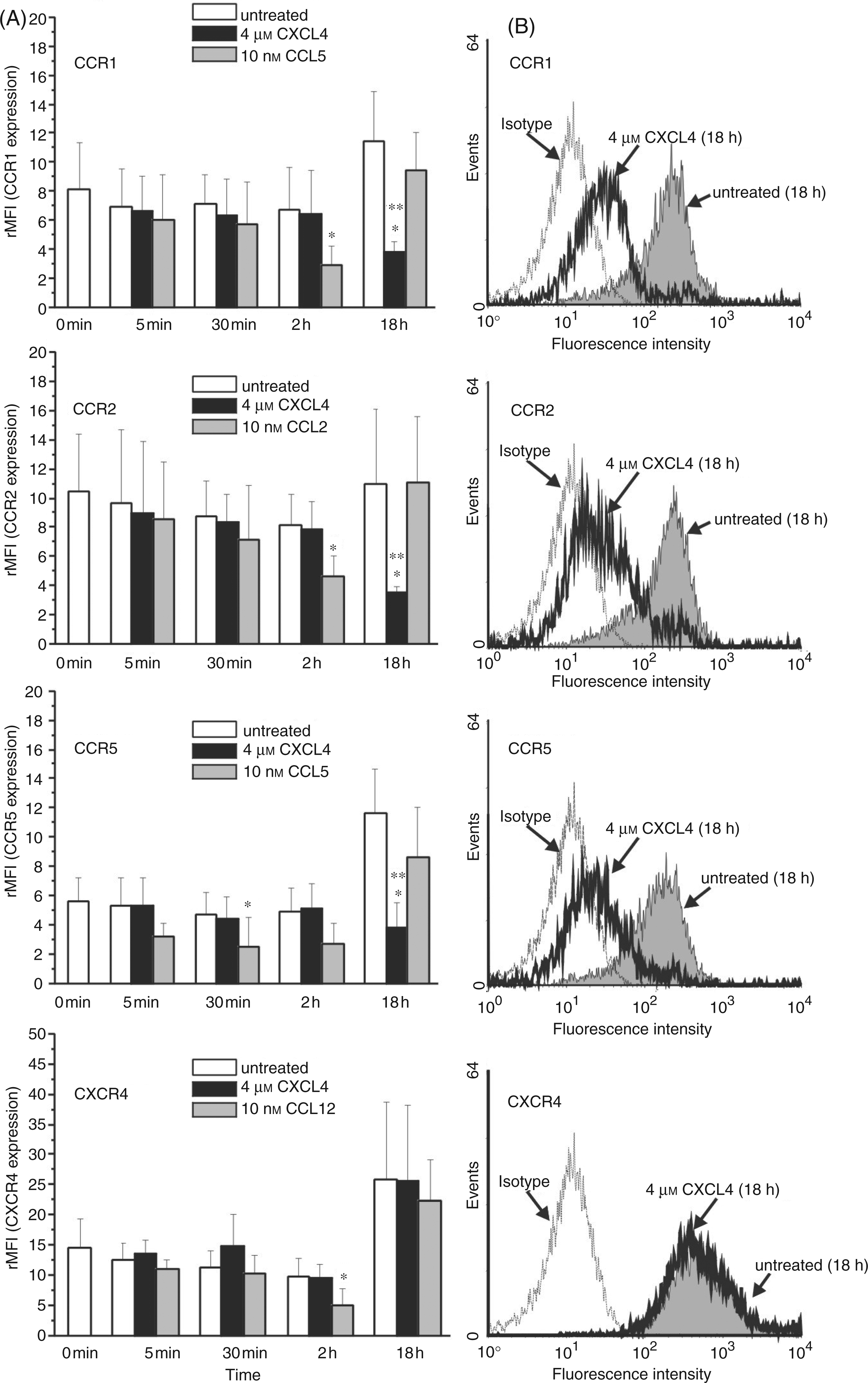
In a second approach, we determined the concentration dependency for CXCL4-mediated CCR1, CCR2, and CCR5 down-modulation by performing dose-response experiments using 0.1–8 µ Effect of CXCL4 dosage on monocyte CC chemokine receptor surface expression. Monocytes (106/ml) were incubated for 18 h in the absence or presence of increasing concentrations of CXCL4 or left unexposed. Subsequently, cells were washed and CC chemokine receptor expression was analyzed by flow cytometry using monoclonal antibodies against CCR1 (upper graph), CCR2 (median graph) and CCR5 (lower graph). Data are derived from four independent experiments. *P < 0.05; cells treated with increasing concentration of CXCL4 versus cells left untreated for the same time period.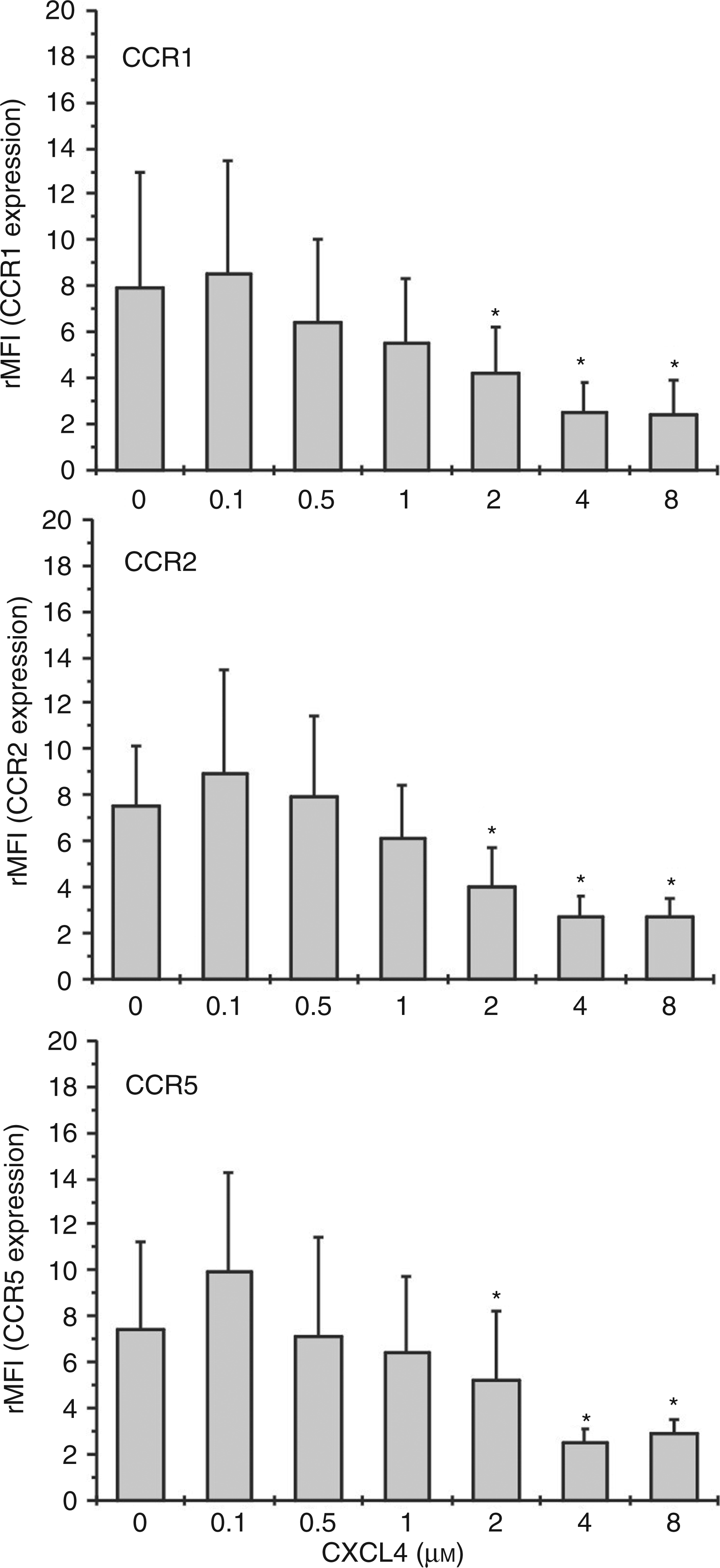
Effect of CXCL4 on CCR1-, CCR2- and CCR5-ligand-induced chemotaxis of human monocytes
Having seen CXCL4-induced down-regulation of CCR1, CCR2 and CCR5 on monocytes, we next examined whether the decrease in surface expression of these receptors would coincide with a change in the cells’ chemotactic responsiveness to their known activating ligands. To verify this, monocytes, incubated with 4 µ Impact of CXCL4 on monocyte chemotaxis induced by CCR1, CCR2, CCR5 and CXCR4 ligands. Monocytes (106/ml) were cultured for 18 h in the presence (circles) or absence (squares) of 4 µ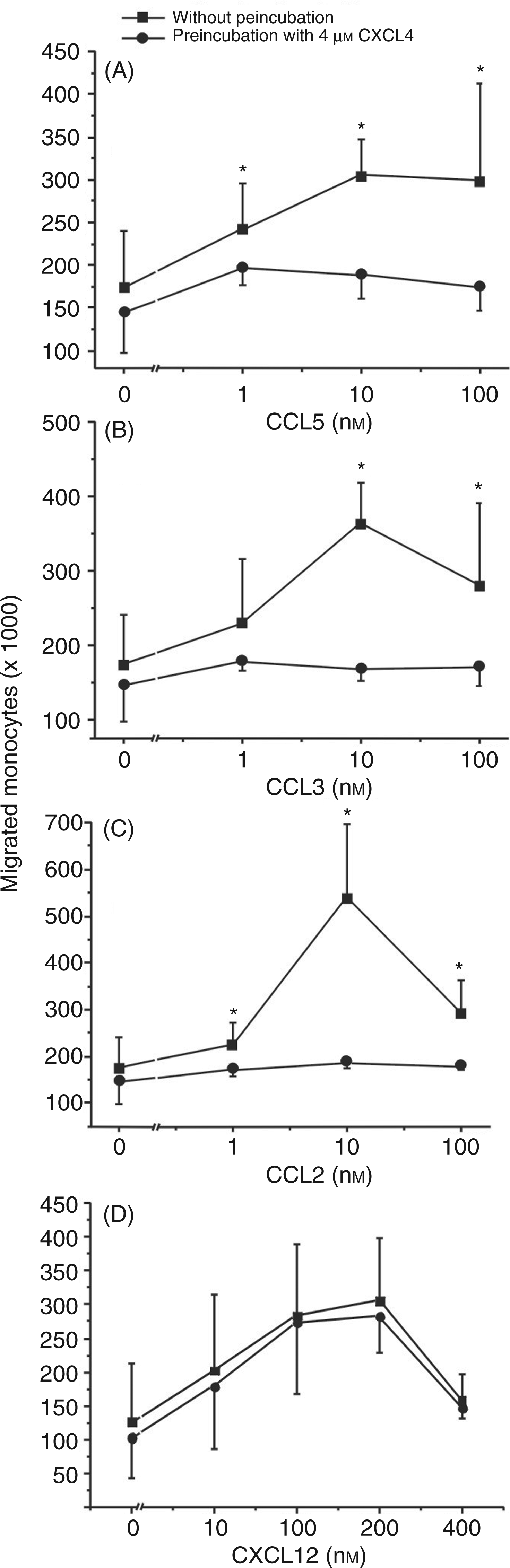
Involvement of TNF-α but not IL-1 in CXCL4-induced CCR1, CCR2 and CCR5 modulation
Down-regulation of chemokine receptors by their specific, homologous ligands (homologous desensitization) has been described as a rapid process limiting pro-inflammatory responses of the target cell.28,29 By contrast, receptor down-regulation by CXCL4 in our experiments, occurring only after 18 h, proceeded rather slowly and suggested that CXCL4 might down-regulate receptor expression and desensitize monocyte chemotaxis via indirect mechanisms, e.g. by stimulating the release of secondary mediators acting in an autocrine manner. It has been shown that CXCL4 can induce TNF-α release in monocytes and, moreover, that exogenous TNF-α may decrease CCR5 expression in monocytes and peripheral macrophages.14,30 In fact, we found that monocytes stimulated with 4 µ Modulation of monocyte CC-chemokine receptor expression by exogenous TNF-α and effect of neutralizing anti-TNF-α antibody on CXCL4-induced CCR1, CCR2 and CCR5 down-regulation. Monocytes (106/ml) were incubated for 18 h in the absence or presence of 4 µ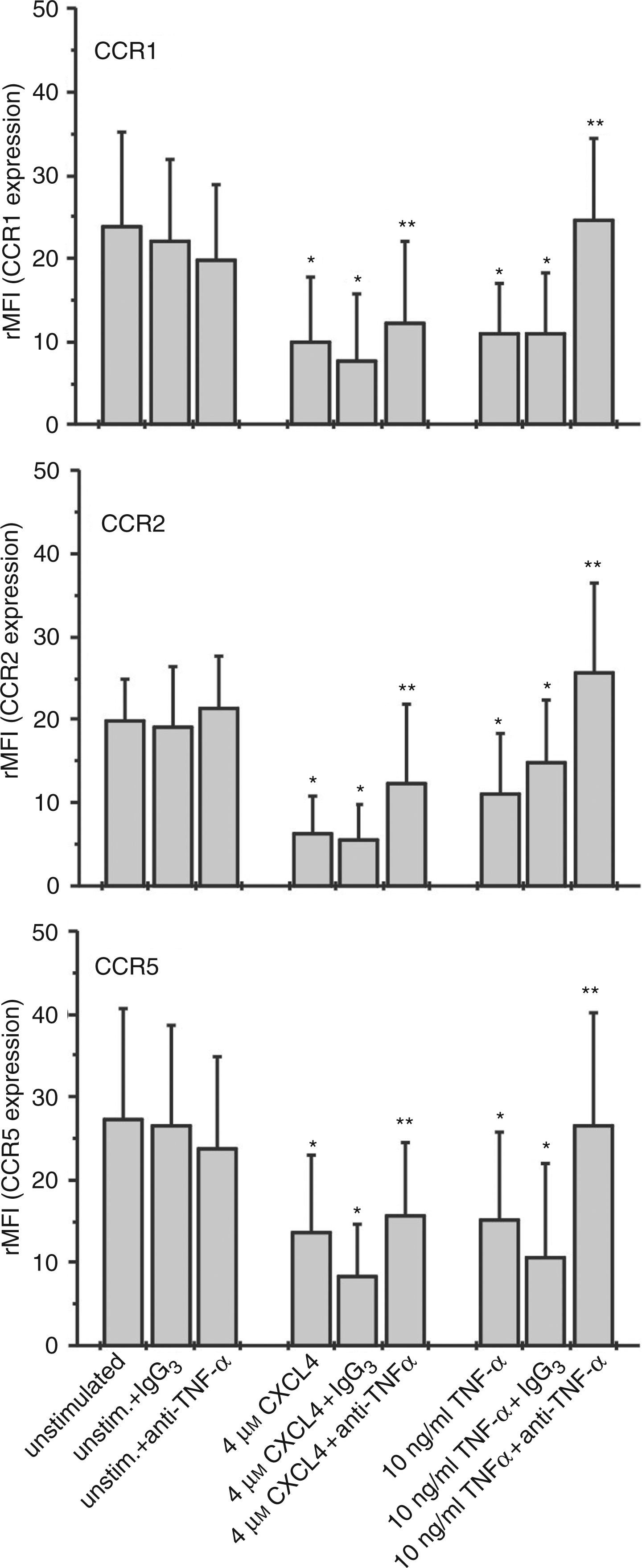
Involvement of TNF-α in CXCL4-induced CC and CXC chemokine release in human monocytes
Having seen that TNF-α is involved in CXCL4-mediated CC-receptor down-regulation, and taking into account that TNF-α is known to induce a variety of chemokines in monocytes, we asked the question, whether TNF-α-stimulated release of corresponding CC chemokine ligands could be responsible for receptor down-regulation.30,32–
36
To investigate this, monocytes were cultured in the presence or absence of CXCL4 (Fig. 5A–D,I) or with TNF-α (Fig. 5E–I) for increasing periods of time, and cell-free culture supernatants were tested for the presence of released chemokines representing ligands for CCR1 (CCL3), CCR2 (CCL2), and CCR5 (CCL3, –4, –5) by ELISA. For a control, the secretion of CXCL8 was also monitored. As expected, CXCL4 as well as TNF-α-challenged monocytes not only released high amounts of CXCL8 but, in addition, also secreted CCL2, CCL3, and CCL4. Although release of the individual chemokines followed more or less different time kinetics, CXCL4-induced maximal or near maximal levels in supernatants were reached after 10 h (CCL2, CCL3) or 6 h (CCL4) and persisted (CCL2, CCL3) or moderately decreased (CCL4) at least until 18 h post stimulation. Interestingly, CXCL4 stimulation did not induce detectable CCL5 release for up to 72 h, while TNF-α, as described by others, induced CCL5 release starting relatively late at about 18 h with a maximum release after 36 h of stimulation.30,32 Taken together, these results clearly show, that CXCL4 as well as TNF-α are able to induce CCR1, CCR2 and CCR5 ligand secretion in human monocytes within a time frame allowing receptor down-regulation.
Involvement of TNF-α in CXCL4-induced chemokine release by monocytes. Monocytes (106/ml) were stimulated with 4 µ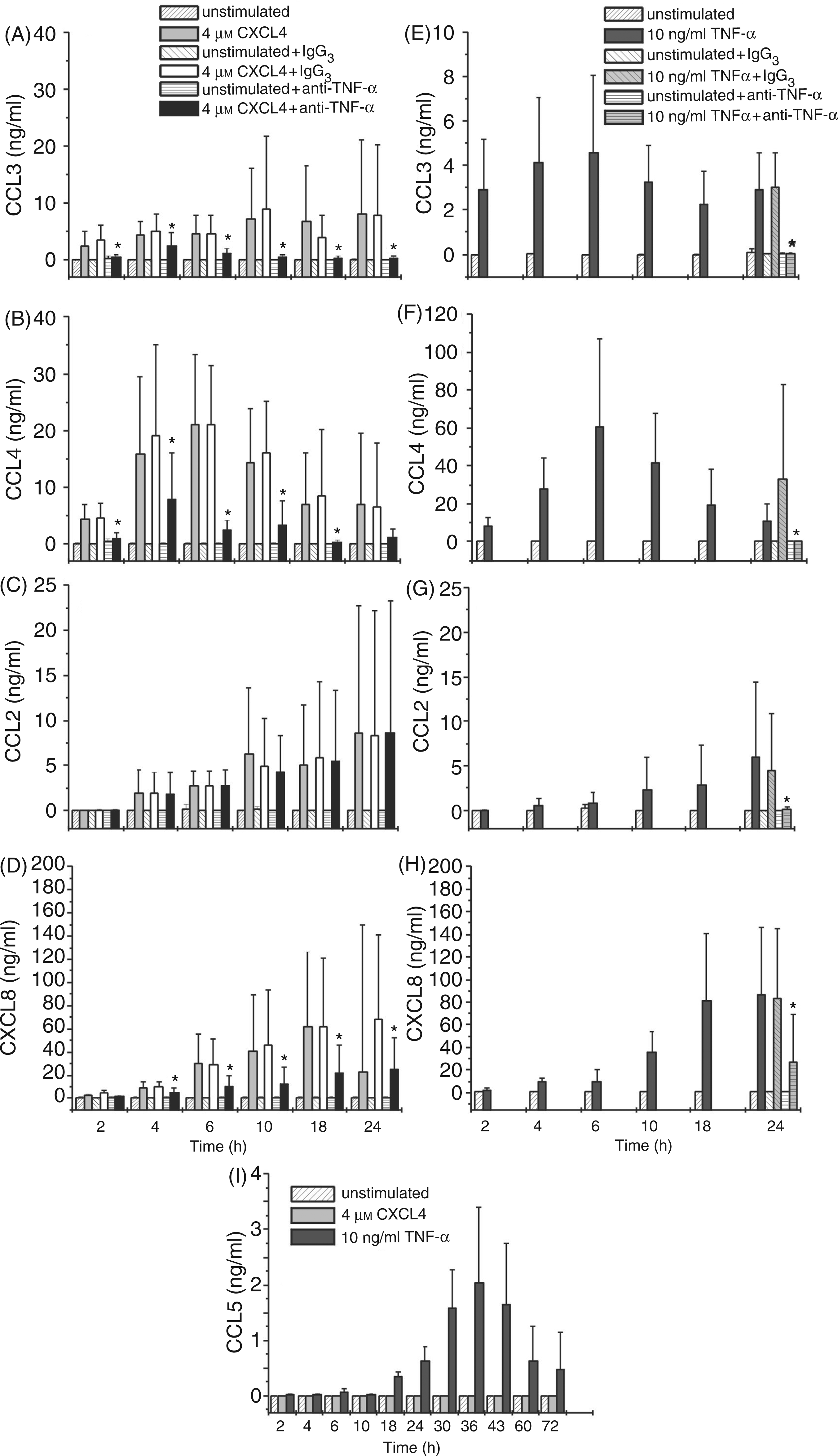
To analyze to what degree TNF-α might be involved in CXCL4-induced chemokine release, CXCL4-challenged monocytes were co-cultured with inhibitory anti-TNF-α antibody or irrelevant antibody of the same isotype (control IgG3) for increasing periods of time and subsequently monitored for their chemokine release during stimulation (Fig. 5A–D). For a control, TNF-α-stimulated cells were co-cultured with the same amounts of inhibitory anti-TNF-α antibody or irrelevant antibody for 24 h (Fig. 5E–H). As expected, exposure of TNF-α-stimulated cells to neutralizing anti-TNF-α drastically reduced the release of all chemokines tested. Thus, in comparison to cells having received irrelevant antibody, CCL3, CCL4, and CCL2 release was almost completely inhibited (by about 98.5 ± 1.8%, 99.0 ± 1.0%, and 97.5 ± 3.1%, respectively) while CXCL8 release appeared reduced by about 76.6 ± 23.9% (Fig. 5E–H). In CXCL4-stimulated cells, release of CCL3, CCL4 and CXCL8 was strongly inhibited over the entire time-course (2–24 h) of culture. Interestingly, CCL2 release remained completely unaffected by the presence of inhibitory anti-TNF-α antibody at all time points tested. Briefly, these data indicate that autocrine TNF-α is involved in CXCL4-mediated release of chemokines CCL3, CCL4 and CXCL8 but not in the release of CCL2. This suggests either direct induction of CCL2 by CXCL4 or by a CXCL4-induced mediator other than TNF-α.
Corresponding results were obtained when examining the involvement of TNF-α in CXCL4-induced mRNA-expression for these chemokines. Following stimulation of monocytes with 4 µ
Relevance of CXCL4-induced chemokines for CCR down-regulation and monocyte chemotactic migration
Next we investigated whether the quantities of chemokines CCL3, CCL4, and CCL2 found in supernatants of CXCL4-stimulated monocytes would be sufficient to down-regulate expression of their specific receptors and/or to stimulate monocytes for chemotaxis. For this, freshly isolated monocytes were exposed to cell-free supernatants harvested from CXCL4-stimulated or unstimulated monocytes and analyzed for expression of CCR1, CCR2, and CCR5 after 2 h. Additionally, monocytes were tested for chemotactic migration towards serial dilutions of the supernatants in a Transwell migration assay. As shown in Figure 6A, supernatants from CXCL4-stimulated monocytes significantly reduced CCR1, CCR2, and CCR5 expression as compared to supernatants from cells cultured for the same time period (10 h) in medium alone. Even more drastic reduction of receptor expression by their ligands CCL2 and CCL5 demonstrated unimpaired functionality of the cells. Corresponding results were obtained when monocyte-derived supernatants were tested for chemotactic activity (Fig. 6B). While serial dilutions of supernatants from CXCL4-stimulated cells stimulated significant concentration-dependent chemotactic migration in freshly isolated monocytes, no migration was obtained in response to supernatants from unstimulated cells. Taken together, these results clearly show that supernatants from CXCL4-stimulated monocytes contain sufficient amounts of chemokines/chemotactic factors to affect monocyte biological function. Attempts to estimate the contribution of chemokines CCL2, CCL3, and CCL4 to receptor down-regulation by adding neutralizing anti-chemokine antibodies to monocytes during CXCL4 stimulation were unsuccessful, because cells exposed to these antibodies produced high unspecific background signals during subsequent flow cytometry analysis of receptor expression. This occurred although normal mouse serum (diluted 1/100) to block unspecific antibody binding was added to cells before the staining procedure (data not shown). Alternatively, we examined whether neutralizing antibodies to the chemokines would affect monocyte migration to CXCL4-induced supernatants. As shown in Figure 6C, addition of anti-CCL2 as well as of anti-CCL3 antibodies significantly reduced chemotactic migration in comparison to an isotype control, while no effect was seen with antibodies to CCL4, and with antibodies to CCL5 and CXCL8 used for control. However, supplementation of supernatant with a combination of all neutralizing antibodies mentioned above lead to somewhat stronger reduction of chemotactic activity, as compared to samples receiving anti-CCL2 or anti-CCL3 alone. These results indicate that CXCL4-induced chemokines CCL2 and CCL3 are involved in stimulating monocyte chemotactic migration and thus probably also in down-regulation of their monocyte-expressed specific receptors. However, because neutralizing these chemokines resulted in only partial inhibition of supernatant-induced chemotaxis, it appeared that additional chemokines/chemotactic factors contributed to monocyte migration and, consequently, receptor down-regulation.
Impact of CXCL4-induced culture supernatants on CCR expression and chemotactic activity in monocytes. Monocytes (106/ml) were cultured for 10 h in the presence or absence of 4 µ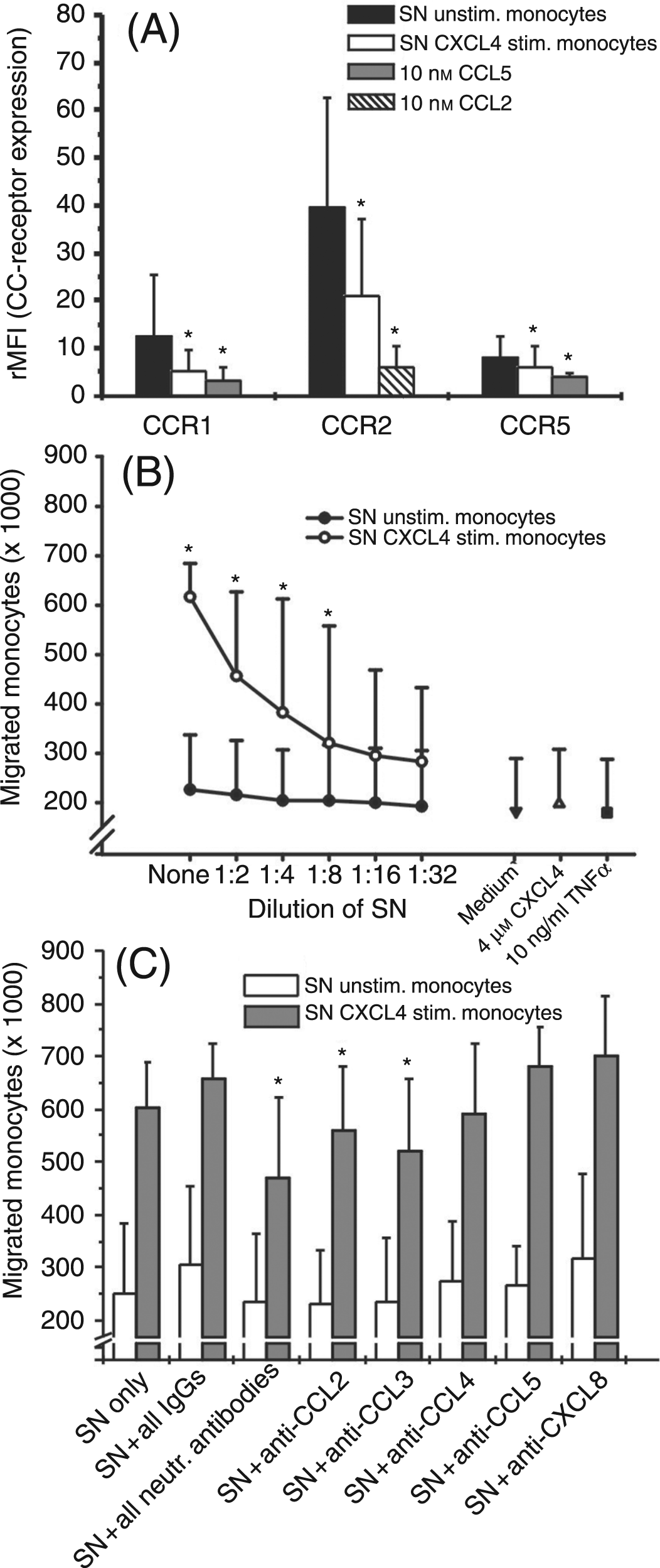
In a final attempt to estimate the importance of CCL2, CCL3, and CCL4 for CCR down-regulation, we used monoclonal antibodies specific for these chemokines to immunodeplete CXCL4-induced monocyte supernatants. For this, we incubated CXCL4-induced monocyte supernatants as well as supernatants from cells left unstimulated for 10 h with a mixture of antibodies to CCL2, CCL3, and CCL4 and with control antibodies of the corresponding isotypes in parallel. As shown in Figure 7 by a representative experiment out of four, incubation of freshly isolated monocytes with CXCL4-induced supernatant reduced CCR expression (CCR1 from rMFI = 15.6 ± 5.1 to 5.4 ± 2.4; CCR2 from rMFI = 13.9 ± 10.8 to 5.9 ± 2.6, and CCR5 from rMFI = 3.4 ± 0.9 to 1.7 ± 0.3). Incubation with CXCL4-induced supernatant having been treated with control isotype antibodies resulted in down-regulation to practically identical degrees (CCR1 to rMFI = 6.1 ± 2.8, CCR2 to rMFI = 7.7 ± 4.6, and CCR5 to rMFI = 1.7 ± 0.3), while pretreatment of supernatant with specific chemokine antibodies partially prevented CCR1 down-regulation (rMFI = 11.2 ± 6.4) and abrogated down-regulation of CCR2 (rMFI = 15.4 ± 10.4) and CCR5 (rMFI = 3.1 ± 0.2). These results clearly show that chemokines CCL3, CCL4, and CCL5 are responsible for, or at least contribute to, CXCL4-mediated CCR1, CCR2 and CCR5 down-regulation.
Immunodepletion of CXCL4-induced monocyte culture supernatants with anti-CCL2, CCL3, and CCL5 antibodies prevents down-regulation of CCR expression. Cell-free supernatants (SN) of CXCL4-stimulated and unstimulated monocytes were harvested after 10 h of culture and either supplemented with a mixture of antibodies to CCL2, CCL3 and CCL4 or a mixture of the same amount of antibodies of the corresponding isotypes for 45 min. Following absorption with Protein A-Sepharose for 30 min and clearance by centrifugation, concentration and reconstitution in PBS/0.1% BSA to volumes corresponding to those of the original supernatant, supernatants or buffer were added for 2 h to freshly isolated monocytes. Subsequently cells were washed and incubated with PE-labeled monoclonal antibodies against CCR1 (top), CCR2 (middle) and CCR5 (bottom) or corresponding isotype controls on ice and analyzed for receptor expression by flow cytometry. Data are derived from one representative out of four independent experiments.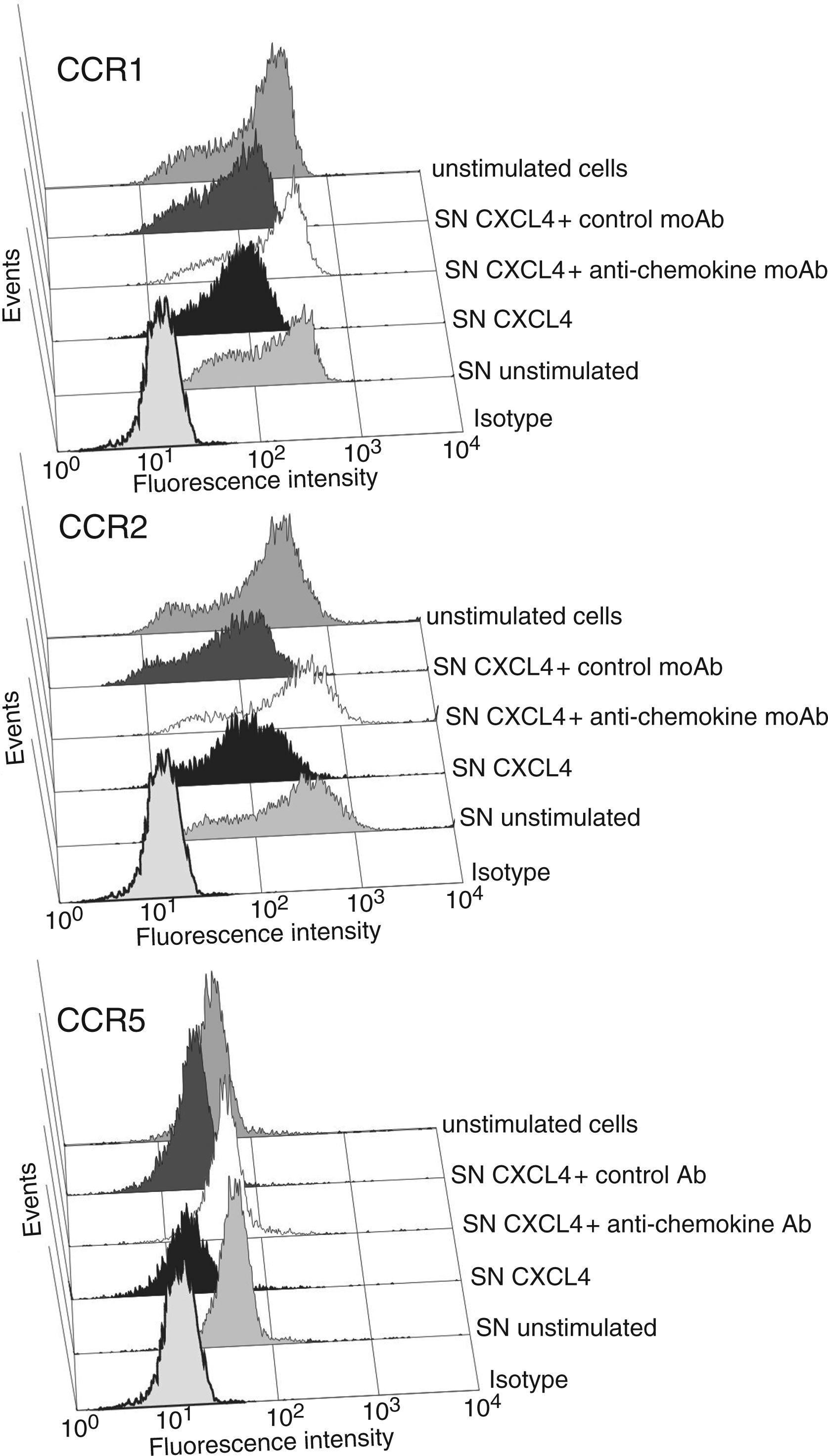
Discussion
The platelet-derived chemokine CXCL4 has been shown to represent a powerful activator of monocyte antimicrobial effector functions such as oxygen radical formation and phagocytosis, whereas it lacks the ability to chemotactically activate and recruit these cells to sites of inflammation.8,31 As has been described by others previously, CXCL4’s contribution to monocyte recruitment may rather be indirect, e.g. by inducing the secretion of various monocyte-directed chemokines or by enhancing CCL5-induced monocyte arrest to endothelium through heterophilic interaction with the latter chemokine.20,31,37 In the present study, we show for the first time that CXCL4 may also interfere with monocyte chemotactic migration by down-regulating the surface expression of several chemokine receptors in these cells. As a consequence, exposure of monocytes to CXCL4 resulted in drastically reduced chemotactic responsiveness to CCR ligands CCL2, CCL3 and CCL5, whereas migration in response to the sole CXCR4 ligand CXCL12 remained completely intact. CXCL4-mediated CCR down-regulation was concentration-dependent, starting upon incubation with 2 µ
As a remarkable dynamic characteristic, CCR down-regulation in response to CXCL4 occurred after a considerably longer time-period of pre-incubation (18 h) as compared to that (30 min to 2 h) observed in response to their cognate chemokine ligands (CCL2, CCL4, CCL5). This phenomenon indicated that CXCL4, which is not a ligand for CCR1, CCR2, or CCR5, exhibited its effect in a way different from direct ligand-induced receptor desensitization/internalization. So far, only few examples of heterologous cross-regulation among chemokines have been described, e.g. for CXCL8 which was found to cross-desensitize receptors CCR5 and CXCR4 on monocytes, resulting in reduced responsiveness toward their specific ligands CCL5 and CXCL12.39,40 Similarly, others reported that ligands of CXCR4 and CCR5 mutually desensitized adhesive and chemotactic responses in human T-cells. 41 However, in either case, the authors reported that desensitization occurred within minutes and was accompanied by rapid intracellular phosphorylation events, while CXCL4-mediated CCR down-regulation in our setting was dependent on the induction and secretion of cytokines and chemokines as second mediators, thus requiring much more time to become established.
One reason for this might be that monocytes do not express a GPCR for CXCL4 as these cells do for most other chemokines, but that signals in response to CXCL4 appear to be transmitted upon binding to a cell-surface chondroitin sulphate proteoglycan, as has already been shown for neutrophils.16,17 In fact, there are many reports describing the modulation of chemokine receptor expression on monocytes/macrophages via non-GPCR ligands, including many cytokines and growth factors. 40 For example, IFN-γ has been shown to inhibit CCR2 expression on human monocytes and IL-13 and GM-CSF to decrease CCR5 expression in human macrophages.42–44 As a cytokine with pleiotropic functions, also TNF-α has the capacity to modulate chemokine receptors in monocytes/macrophages, as exemplified by its ability to reduce the expression of CCR5 and CCR2, correlating with decreased HIV replication and CCL2-mediated monocyte endothelial transmigration, respectively.30,45 Because TNF-α is one of the cytokines that are induced by stimulation with CXCL4 in monocytes, we hypothesized that it could represent one of the secondary mediators responsible for CCR down-regulation in these cells. 14 In agreement with the authors cited above, we found that externally applied TNF-α reduced expression of CCR2 and CCR5 in monocytes, and in addition also down-regulated CCR1 to a similar extent. The involvement of autocrine TNF-α in CXCL4-induction became clear by our findings that neutralizing anti-TNF-α antibody strongly prevented down-regulation of all three CCRs. However, the fact that even saturating dosages of anti-TNF-α only partially abrogated this effect strongly suggested the participation of other secondary mediators.
Interestingly, much, if not all, of the effect by autocrine TNF-α on CCR down-regulation was also indirect, being due to the cytokine’s capacity to induce the synthesis of various chemokines in monocytes. Thus, neutralizing antibody to TNF-α not only inhibited the secretion of CXCL8 but also that of CCR1/CCR5 ligands CCL3 and CCL4 by CXCL4-stimulated monocytes, suggesting that these chemokines were not directly induced by CXCL4 but required TNF-α as a secondary mediator. Surprisingly, anti-TNF-α antibody was without effect on the CXCL4-induced secretion of CCR2 ligand CCL2, although TNF-α as a primary stimulus was able to induce CCL2 secretion. This indicates that there may exist different ways of CCL2-induction in CXCL4-stimulated monocytes with TNF-α having only a minor contribution. Irrespective of these considerations it appears, however, clear that CXCL4/TNF-α-induced and secreted chemokines contribute to CCR down-regulation, as treatment of culture supernatants from stimulated cells with neutralizing antibodies to CCL2 or CCL3 significantly reduced monocyte chemotactic migration towards these supernatants. These results are very similar to those reported previously for the down-regulation of CCR1, CCR2, and CCR5 by C-reactive protein (CRP) on human adherent monocytes which was a result of chemokine production, namely of CCL2, CCL3, and CCL4 by these cells. 46 Although, in our experiments, a combination of various anti-chemokine antibodies was somewhat more effective in reducing monocyte chemotaxis as compared to every antibody alone, their effect was far from being complete (see Fig. 6C). This suggests that still other chemokines or chemotactic substances participate in CCR down-regulation. Nevertheless, by specifically immunodepleting CCL2, CCL3 and CCL4 from monocyte culture supernatants, we got evidence that the latter three chemokines play major roles in CXCL4-mediated CCR down-regulation. Although immunodepletion consistently abrogated the down-regulatory capacity in CXCL4-induced supernatants, as seen by strong reduction of CCR1 down-regulation and complete prevention of CCR2 and CCR5 down-regulation, it would, however, be premature to conclude that the above chemokines are necessarily the only ones responsible for CCR down-regulation. Because synergistic actions between different chemokines are a common phenomenon, one could imagine that, in addition to CCL2, CCL3, and CCL4, other chemokines interacting with CCR1, CCR2, and CCR5 participate in down-regulating these receptors. Potential candidates could be CCL7, a ligand for CCR1 and CCR2, as well as CCL8, a ligand for CCR2 and CCR5. Most interestingly, it was found that monocytes also secrete heterodimers of CCL3/CCL4 that could impact CCR5 and possibly other CCR functions. 47
Changes in CCR expression have also been found to represent a characteristic feature during the differentiation of monocytes into macrophages. In previous studies, we could demonstrate that CXCL4 represents a survival and differentiation factor for monocytes, generating a special type of macrophage with decreased expression of HLA-DR and enhanced capacity for unspecific phagocytosis.8,14 Interestingly, as a result of in vitro differentiation for 6 d, CXCL4-exposed macrophages expressed considerably less surface CCR5 as compared to M-CSF-exposed cells. 48 By contrast, differentiation of monocytes in response to other agents such as autologous serum was shown to result in the up-regulation of CCR1 and CCR5, while CCR2 underwent down-regulation. 49
Thus, it appears that CXCL4 generates a special type of macrophage characterized by loss of reactivity to inflammatory CC chemokines due to CCR down-regulation. The fact that this process becomes effective relatively early during monocyte differentiation (after 18 h) may be an adaptation to conditions found at sites of platelet activation. At such inflammatory sites, recruitment of monocytes is likely to be initiated by platelet-released CC chemokines (predominantly CCL5), while simultaneous stimulation of these monocytes by CXCL4 leading to production of inflammatory chemokines CCL2, CCL3, CCL4, and still others may serve to induce a retarded second wave of monocyte recruitment. Down-regulation of CCR as a consequence of the receptors interaction with their chemokine ligands additionally may be instrumental in retaining the cells at the inflammatory site. Conditions within a thrombus are likely to facilitate these kinds of interaction. Enclosed in such sites where platelets and leukocytes are found in close physical association, monocytes will be exposed to platelet-released CXCL4 as well as endogenously produced chemokines for longer time-periods, while binding of chemokines to tissue-expressed glycosaminoglycans will result in high local concentrations of these mediators. These mechanisms are likely to contribute to defence against infection and at the same time might promote wound healing.
Footnotes
Acknowledgements
The authors gratefully acknowledge the supply of platelet concentrates by Dr Gesa Washington and Sonja Schottstaedt (Department of Transfusions Medicine, Universtätsklinikum Schleswig-Holstein, Campus Kiel, Kiel, Germany). The authors thank Dr Kathleen A. Clouse (FDA, CDER/OBP/DMA, Maryland, USA) for technical support by her laboratory and Dr Holger Heine and Dr Oliver Umland (Immunology and Cell Biology, Research Center Borstel, Borstel, Germany) for their introduction to the light cycler technique. We thank Christine Engellenner, Gabriele Huss, Erika Kaltenhäuser and Renate Bergmann for perfect technical assistance. This work was supported, in part, by Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 367, Projekt C4.