
Abstract
In macrophages, mitogen-activated protein kinases (MAPK) are critical regulators of both, mycobacterial replication and mycobacteria-induced cytokine formation. To segregate direct effects of MAPK function on mycobacterial replication from indirect, cytokine-mediated effects, we studied the growth of Mycobacterium avium strains in wild-type and tumor necrosis factor (TNF)-α- or interleukin (IL)-10-deficient bone marrow-derived murine macrophages. Using specific inhibitors of the p38- and the ERK1/2-MAPK pathways, we found that the use of SB203580 always reduced, whereas the presence of PD98059 always promoted, bacterial replication of highly virulent and intermediately virulent M. avium strains, independent of endogenous TNF-α or IL-10. The exogenous addition of TNF-α to TNF-α-deficient and wild-type M. avium-infected macrophages overrode the replication-reducing effect of SB203580, but not the replication-promoting effect of PD98059. In summary, our data demonstrate that a proper balance of MAPK activity is essential for macrophage control of M. avium growth, and that the ratio of the cytokines TNF-α and IL-10 can additionally modulate replication. Our findings indicate a novel therapeutic avenue for treating mycobacterial infections in particular by stimulating ERK1/2 or activating ERK1/2-dependent mechanisms in infected macrophages.
Keywords
Introduction
Mycobacterium avium is the causative agent of a serious, and often disseminated, opportunistic infection in late stages of the acquired immunodeficiency syndrome (AIDS), but also causes localized disease in patients with other immunodeficiencies or even seemingly immunocompetent individuals. Infections with M. avium are difficult to treat because of the primary resistance of this pathogen to many antimicrobial agents. 1 In addition, M. avium infection has frequently been used as an experimental system in vitro to uncover mycobacteria-induced signalling pathways in macrophages, 2 and in vivo to determine mechanisms of mycobacteria-induced tissue pathology. 3
Macrophages often form the first line of defense against pathogens, and in concert with NK-cells and T-cells and inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-12 and interferon (IFN)-γ, are critical in restricting mycobacterial growth. 4 – 6 Macrophages are also the producers of IL-10, one of the principal cytokines counteracting excessive activation and inflammation during antimicrobial defense. 7 Macrophages, paradoxically, are also the preferred habitat for mycobacteria. Mycobacterium tuberculosis and M. avium have evolved mechanisms to persist and replicate in macrophages chronically, because they can inhibit lysosomal acidification and phagolysosomal fusion6,8 or major histocompatibility complex (MHC) class II antigen processing and presentation 9 – protein antigens are degraded into peptides in intracellular vesicles, bound to MHC class II molecules, transported to the cell surface and recognized by CD4 T-cells – and can modify by special cell wall components the production of cytokines such as TNF-α which is necessary for granuloma formation and antimycobacterial activities 6 of macrophages or of IL-10 which modulates macrophage functions. 7
The balance between the bacteriocidal mechanisms of macrophages and the evasion strategies of mycobacteria is, to a significant extent, influenced by the virulence of the infecting strain. For example, the highly virulent M. avium strain TMC724, an isolate derived from infected fowl, replicated progressively in mice and caused expanding, tumor-like infiltrations of macrophages in infected organs. In contrast, the intermediately virulent strain M. avium SE01, an isolate derived from an AIDS patient, induced well-organized granulomas which contained mycobacterial replication at a plateau level. 10 In human and murine macrophages, highly virulent M. avium induced less production of TNF-α and IL-10 than intermediately virulent or apathogenic mycobacteria,11,12 indicating a close link between virulence and immune cell activation.
Intracellular signalling cascades connect the primary interaction of mycobacteria with membrane-localized cellular receptors to a transcriptional and translational response program. The evolutionarily conserved group of mitogen-activated protein kinases (MAPKs) are involved in critical signalling cascades of all eukaryotic cells, such as survival, programmed cell death, differentiation, motility or the production of cytokines.13,14 Four serine–threonine protein kinases have been identified in this context: the extracellular kinase ERK1/2, the MAPK p38, the c-Jun NH2-terminal kinase JNK and the extracellular regulated kinase ERK5. Mitogen-activated protein kinases are activated by specific MAPK kinases, e.g. ERK1/2 by MKK1/2 or p38 by MKK3/6.13,15
Interestingly, it is again the virulence of M. avium strains that determines the extent of MAPK activation. Highly pathogenic isolates cause little activation of p38 and ERK1/2, whereas low or intermediately virulent strains are potent activators.12,16– 18 Specific inhibitors of MAPK cascades, such as SB203580 and PD98059, have been instrumental in uncovering the significance of individual MAPK for the physiological roles of the cell signalling pathways that they inhibit, e.g. the cytokine production by macrophages. The inhibitor SB203580 inhibits the activation of p38 and PD98059 blocks the activation of MKK1/2 and thus of ERK1/2.19,20 Experiments with human and murine macrophages infected with mycobacteria revealed ERK1/2 and, to a certain extent, p38 to be involved in TNF-α production, whereas only p38 was critical for production of IL-10.12,17,21– 23
In a separate approach, three groups have investigated the role of p38 and ERK1/2 for replication of mycobacteria in macrophages. Inhibition of ERK1/2 caused increased replication of M. avium 2151 SmT and M. avium SE01, but not M. avium 2151 SmO, in human macrophages and of M. avium 2151 SmO in mouse macrophages.12,17 In contrast, inhibition of p38 was followed by decreased replication of M. avium 2151 SmT in mouse macrophages, 17 however had no effect on mycobacterial replication in human macrophages. 12 Similar effects have been observed recently with regard to the replication of M. avium intracellulare in human monocytes. 24
To date, no attempt has been made to find a causal connection between these findings. Thus, the critical question if the replication of M. avium is directly influenced via ERK1/2 and p38 or indirectly via the products of MAPK activation, in particular the cytokines TNF-α and IL-10, remains unresolved.
To differentiate between these two possibilities, we performed infection experiments with two M. avium isolates previously characterized to differ in their virulence (SE01 and TMC724) in the presence or absence of MAPK inhibitors in wild-type macrophages, and in macrophages deficient for TNF-α or IL-10 production. Both, cytokine production and intracellular replication of M. avium, were recorded simultaneously. In addition, recombinant murine TNF-α and IL-10 were added to the experimental setting to differentiate better between cytokine and/or MAPK dependent and independent effects.
We now show that replication of M. avium in macrophages is directly influenced by MAPK activity independently of TNF-α and IL-10. More specifically, our data suggest that modifying ERK1/2 activity may be a novel strategy for inhibiting M. avium growth in macrophages.
Materials and methods
Mice and generation of macrophages
Specific pathogen-free wild-type mice (C57BL/6 and C57BL/10; Charles River Wiga GmbH, Sulzfeld, Germany), TNF-α gene deficient mice (C57BL/6-TNF-α−/−), 25 kindly provided by Prof. Dr D. Schlüter (Magdeburg, Germany) and IL-10 gene deficient mice (C57BL/10-IL-10−/−, B10.129P2(B6)-Il10<tm1Cgn> Stock#002250, Charles River) were maintained under barrier conditions at the Research Center Borstel. In all experiments, mice were matched for age, sex and genetic background. For the generation of bone marrow-derived macrophages, marrow cells were obtained by rinsing of femora and tibia. Cells were centrifuged for 10 min at 249 g at 4°C and subsequently resuspended in macrophage culture medium (DMEM with 4.5 g/l glucose [PAA, Cölbe, Germany] containing 10% [v/v] heat-inactivated fetal calf serum [FCS; Biochrom, Berlin, Germany], 2 mm L-glutamine, 1 mm sodium pyruvate, 10 mm HEPES buffer (Invitrogen, Karlsruhe, Germany)) and 50 ng/ml of recombinant human macrophage colony-stimulating factor (rhM-CSF; R&D Systems, Wiesbaden, Germany) at 4°C. Cells were first cultured on a Nunclon™ Surface dish (92 × 17 mm; Nunc, Wiesbaden, Germany). After 24 h of cultivation, non-adherent cells were harvested. Cells were transferred into 10 ml fresh medium with 50 ng/ml rhM-CSF to each culture dish (92 × 16 mm; Nunc) for 7 d at a density of 1.2 × 106 cells/ml. After 3 d, medium with 50 ng/ml rhM-SCF was renewed. On day 7, adherent cells were washed in PBS (Invitrogen), subsequently detached by incubating them for 10 min (37°C, 5% CO2) in Accutase™ (PAA) and, after centrifugation, resuspended in macrophage culture medium in the absence of rhM-CSF. Sacrifice of mice for preparation of cells was approved by the Ministry of Agriculture, the Environment and Rural Areas, Kiel, Germany.
Bacteria
Mycobacterial strains M. avium SE01, 10 and TMC724, obtained from Dr F.M. Collins (Trudeau Institute, Saranac Lake, NY, USA), were grown in Middlebrook 7H9 medium (Difco, Detroit, MI, USA) containing 10% OADC (oleic acid, albumin, dextrose, catalase; BD Biosciences, Heidelberg, Germany) and 0.05% Tween 80 (Sigma, Steinheim, Germany) at 37°C. Mid-log phase cultures were harvested, and aliquots were frozen at –80°C until use. For in vitro experiments, bacterial aliquots were thawed, centrifuged for 10 min at 835 g, resuspended in PBS and bacterial aggregates dispersed by sonication at 35 kHz for 5 min (Sonorex RK 52, Bandelin Electronic, Berlin, Germany). Bacterial lipopolysaccharide (LPS) of Salmonella enterica sv. Friedenau H909 was kindly provided by Prof. Dr H. Brade (Research Center Borstel, Germany).
In vitro infection
For in vitro infection, 3.5 × 105 murine macrophages were seeded in 0.3 ml macrophage culture medium in 48-well flat-bottom Nunclon™ Surface plates (Nunc), cultured (37°C, 5% CO2) overnight (18 h) and infected with 1.05 × 106 CFU of M. avium SE01 or TMC724. Incubation with 10 µm SB203580 (4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazol) or PD98059 (2′-amino-3′-methoxyflavon) (both in DMSO; Calbiochem, Darmstadt, Germany) and as solvent control 0.1% (v/v) dimethylsulfoxide (DMSO, same concentration as in inhibited cultures; Sigma) started 1 h before infection. The incubation of macrophages with cytokines started at the time of infection with 50 ng/ml murine TNF-α (PeproTech, Frankfurt aM, Germany) or with 10 ng/ml murine IL-10 in Tris-HCl (PeproTech) and as solvent control 50 µm Tris (in Tris/HCl; Serva, Heidelberg, Germany). As control stimulus, 10 ng/ml LPS was added to cultures at the time of infection. For determination of MAPK phosphorylation, 4 × 105 murine macrophages were cultured in 0.5 ml macrophage culture medium and infected with 4 × 106 CFU of M. avium SE01 or TMC724 in 24-well flat-bottom Nunclon™ Surface plates (Nunc).
Analysis of mycobacterial growth
Four hours post infection (p.i.) and cultivation (37°C, 5% CO2), non-phagocytosed bacteria were removed by washing three times with 0.3 ml Hanks’ balanced salt solution (HBSS; Invitrogen) at 37°C. After washing and after 3 d of cultivation, 0.3 ml macrophage culture medium at 37°C with inhibitors, cytokines and controls at the initial concentrations were added for 7-d cultivation (37°C, 5% CO2). Tumor necrosis factor-α was added daily at the initial concentration over all 7 d of culture. Prior to CFU determination (4 h, 3 d and 7 d p.i.), cultures were washed three times with 0.3 ml HBSS at 37°C. Subsequently, 0.3 ml macrophage culture medium containing 0.033% Syto®24 (green fluorescent nucleic acid stain, in DMSO; MoBiTec, Göttingen, Germany) for 30 min was added and the fluorescence intensity measured in a microplate reader ((Tecan Genion, software magellan v2.0, Tecan, Crailsheim, Germany), kindly provided by Prof. Dr S. Bulfone-Paus, Research Center Borstel, Germany, at 535 nm after excitation at 485 nm. Macrophages of these cultures were lysed by 0.02% saponin (Sigma) at 37°C in 15 min. Lysates were serially diluted in 0.05% Tween 80 (Merck, Darmstadt, Germany) and plated twice on 7H10 agar containing sodium pyruvate and sodium glutamate (19 g/l Middlebrook 7H10-Agar, 4 g/l Nutrient Broth; both BD Biosciences; 0.76 g/l sodium pyruvate, 5.5 g/l glucose, 0.5 g/l sodium glutamate, 8 g/l glycine, all from Merck). After 3 weeks at 37°C, the CFUs were counted.
Detection of cytokine release
At indicated times p.i. and cultivation (37°C, 5% CO2), aliquots of culture supernatants were taken and kept at –20°C until further analysis. The samples were analyzed for TNF-α and IL-10 levels by ELISA as recommended by the manufacturer (Duo Set® Mouse TNF-α, R&D systems; BD OptEIA™ Set Mouse IL-10, BD Biosciences).
Determination of MAP kinase phosphorylation
At indicated times p.i. and cultivation (37°C, 5% CO2), cells were lysed by adding 200 µl sample buffer (pH 6.8, 62.5 mm Tris-HCl, 80 mm dithiothreitol (DTT), 10% (v/v) glycerol; all Merck; 2% (w/v) sodium dodecyl sulfate (SDS); Amersham, Braunschweig, Germany; 0.002% (w/v) bromophenol blue; Serva, Heidelberg, Germany) and stored at –20°C. After incubation at 95°C for 5 min and centrifugation at 13,000 g and 20°C for 5 min, equal amounts of protein were loaded onto a 12% SDS-polyacrylamide gel (SDS-PAGE; M10.1177_1753425910Protean 2 System, BioRad Laboratories, München, Germany). Gel electrophoresis was performed at 70 V for 15 min followed by 200 V for 45 min at room temperature (20–22°C) and subsequently transferred to a nitrocellulose membrane (Sartorius, Göttingen, Germany) for 90 min by wet blot at 75 V, 0.16 A/cm2 and 4°C (M10.1177_1753425910Protean 2 System, BioRad). The membrane was blocked for 1 h with 5% non-fat dry milk (Saliter, Obergünzburg, Germany) in T-TBS (pH 7.2, 285 mosm; Tris-buffered saline (TBS) containing 0.1% (v/v) Tween 20; both Merck), washed three times for 15 min with T-TBS and incubated overnight at 4°C with the 1:1000 in 5% non-fat dry milk (in T-TBS) diluted primary antibody (p38MAPK, phospho-p38MAPK (Thr 180/Tyr 182), p44/42 (ERK1/2)MAPK, phospho-p44/42 (ERK1/2)MAPK (Thr 202/Tyr 204); all antibodies from rabbit; all from Cell Signalling, Frankfurt, Germany). Membranes were washed like the day before, incubated for 1 h with the 1:4000 in 5% non-fat dry milk (in T-TBS) diluted secondary antibody (horseradish peroxidase [HRP]-conjugated goat anti-rabbit IgG; Jackson Immuno Research, Dianova, Hamburg, Germany) and washed three times for 20 min with T-TBS. Immunoreactive bands were developed using a chemiluminescence assay (ECL substrate and ECL hyperfilm, Amersham) as recommended by the manufacturer.
Mathematical and statistical analysis
Detailed statistical analyses were performed for all major issues addressed in the current study. Mean and SD were obtained from a minimum of three independent experiments, each with at least two (analysis of cytokine release, MAPK-phosphorylation, intracellular replication) and up to four (analysis of intracellular replication) technical replicates. Since macrophage cell numbers in infection experiments may be affected by the virulence of the used M. avium strain, added cytokines and the changed activity of intracellular MAPK by treatment with specific inhibitors, we related M. avium CFU to the macrophage numbers as follows. The Syto®24 fluorescence of each technical replicate (e.g. cytokine treated) was divided by the fluorescence mean of the reference replicates with M. avium SE01 infected wild-type macrophages without inhibitors and cytokines, leading to a ‘cell density factor’. So the CFU of a given culture was divided by 350,000 seeded macrophages and subsequently divided by the cell density factor of macrophages of the given culture at day 7. The result is referred to as ‘relative CFU’, which describes the relative number of mycobacteria inside viable, adherent macrophages. Data analysis was performed using Microsoft® Excel (Office 2004 Professional, Microsoft, Redmond, WA, USA) and GraphPad Prism® v5.0 (GraphPad Software Inc., San Diego, CA, USA).
Statistical analysis was done using JMP® v8.0 (SAS Institute Inc., Cary, NC, USA). Prior to analysis, data were log transformed to ensure homoscedasticity. A value of P < 0.05 was always considered significant. The experimental design involved the following fixed factors: time, bacterial strain, inhibitor, macrophage phenotype, addition of cytokine. The experimental design was always completely randomized block design and data were analyzed by mixed model analysis with the experimental day as the blocking variable and, if required (P < 0.05), the control mean values of the experimental days as a co-variate. Subsequently, starting with the highest order interaction terms, non-significant interaction terms were successively excluded from the model. Finally, factors showing significance were analyzed by the Tukey–Kramer test. The statistical model used for analysis is given in the figure captions.
Results
Infection of murine macrophages with M. avium: replication and strain-dependent modulation of the cellular response
Bone marrow-derived macrophages were infected with M. avium SE01 and TMC724 in vitro with a multiplicity of infection (MOI) of three mycobacteria per macrophage. Four hours post infection (p.i.), the uptake of M. avium SE01 and TMC724 into macrophages was similar (Fig. 1A). No significant change in the bacterial burden was observed during the first three days of infection. However, the analysis on day 7 showed a significant, 29-fold increase in colony forming units (CFU) in cultures infected with M. avium SE01 compared to the levels measured 4 h p.i. In infections done with M. avium TMC724, an 8-fold increase in intracellular bacterial load was observed. Thus, both M. avium strains are actively replicating in murine macrophages. The cellular response to infection with the two isolates was strikingly different: M. avium SE01 induced a strong TNF-α release after 5 h of infection, whereas upon infection with M. avium TMC724, hardly any cytokine formation was measurable (Fig. 1B). Similar effects were observed when IL-10 formation was monitored 48 h p.i. (Fig. 1C).
Mycobacterial replication and the cellular response of murine macrophages after infection with M. avium SE01 and TMC724. Murine bone marrow-derived C57BL/6 macrophages were infected with M. avium SE01 and TMC724 with a multiplicity of infection (MOI) of 3 mycobacteria per macrophage. (A) The number of colony forming units (CFUs) per seeded macrophage after 4 h, 3 d and 7 d is shown as mean with SD of independent experiments (n = 10, 4 h; n = 4, 3 d; n = 11, 7 d). In two-factorial analysis (factors: bacterial strain, time) the interaction was highly significant (P = 0.0002); subsequent analysis (Tukey–Kramer) showed that the CFUs at day 7 were higher than at 4 h or 3 d (P < 0.05), and also that the CFUs of the SE01 were higher compared to the TMC724 strain (P < 0.05). (B,C) Cytokine levels in supernatants of M. avium-infected (MOI = 3) macrophage cultures were analysed for TNF-α after 5 h and for IL-10 after 48 h by ELISA. Shown are mean values of duplicates ± SD of one representative experiment out of three independent experiments. (D) Phosphorylation of ERK1/2 and p38 MAPK during the M. avium infection. Bone marrow-derived C57BL/6 macrophages were infected with M. avium SE01 and TMC724 at a MOI of 10 bacteria per macrophage and mitogen activated protein kinase (MAPK) phosphorylation was analyzed at the time points indicated. Cells were lysed, and aliquots of cell lysates were separated by SDS-PAGE and immunoblotted onto nitrocellulose membranes. The membranes were incubated with specific anti-phospho-ERK1/2 and anti-phospho-p38 antibodies and then incubated with a peroxidase-coupled secondary antibody. Visualization was performed by enhanced chemiluminescence. As controls, the amounts of total ERK1/2 and p38 were detected in the same lysates.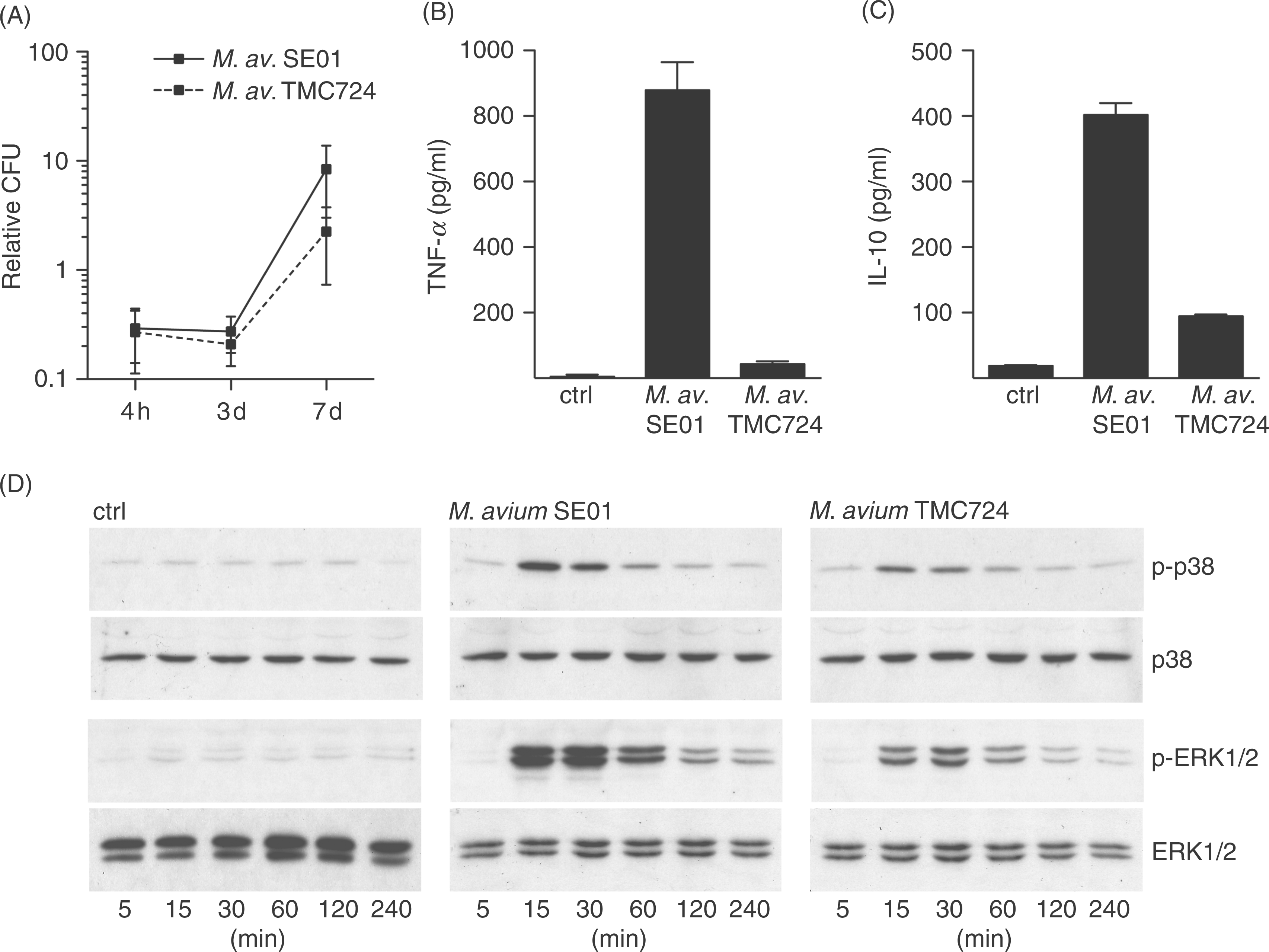
These strain-specific differences were also detectable early on during infection at the level of MAPK activation. Stimulation with M. avium SE01 caused a stronger degree of phosphorylation of the MAPK p38 and ERK1/2 compared to M. avium TMC724 during the first 4 h of infection (Fig. 1D). Unstimulated macrophages (ctrl) did not show an increased phosphorylation of p38 and ERK1/2 during the whole time course. The first and maximal signal for both kinases was detected 15 min after infection with both strains. During the following hours, a continuous decrease of the phosphorylation level in the infected macrophages was observed.
These data indicate that infection with the highly virulent M. avium is associated with a less pronounced host cell activation, when compared to the intermediately virulent M. avium strain.
The MAP-kinase inhibitor SB203580 leads to a decreased, PD98059 to an increased replication of M. avium in macrophages
Macrophages were incubated with 3 µm and 10 µm SB203580 or PD98059 for 1 h prior to and also during the infection with M. avium SE01 or TMC724. After 4 h and 7 d p.i., macrophages were washed, labelled with Syto®24 (see Material and Methods), and the lysates analyzed for CFU. The addition of SB203580 or PD98059 had no effect on the uptake of M. avium SE01 and TMC724 in murine macrophages, when compared to untreated and also solvent control treated cells (DMSO 0.1% [v/v]; Fig. 2A).
The MAPK inhibitors SB203580 and PD98059 regulate replication and cytokine formation of M. avium-infected murine macrophages. Cultures of macrophages were incubated with 3 µm or 10 µm of the MAPK inhibitors SB203580 or PD98059 or 0.1% (v/v) DMSO as solvent control (ctrl) for 1 h before and during infection. Macrophages were infected with M. avium SE01 and TMC724. (A) The CFU counts in lysed macrophages were determined 4 h after infection. Values represent mean ± SD of the relative CFU of five independent experiments. (B) Relative CFU was determined 7 d post infection (p.i.). Values represent means of duplicates ± SD of one representative experiment out of five independent experiments with 10 µm MAPK inhibitor. For definition of relative CFU please see Materials and Methods. (C–F) Cytokine levels in supernatants of M. avium-infected macrophage cultures were analysed for TNF-α and IL-10 at the times indicated by ELISA. Shown are mean values of duplicates ± SD of one representative experiment out of three independent experiments.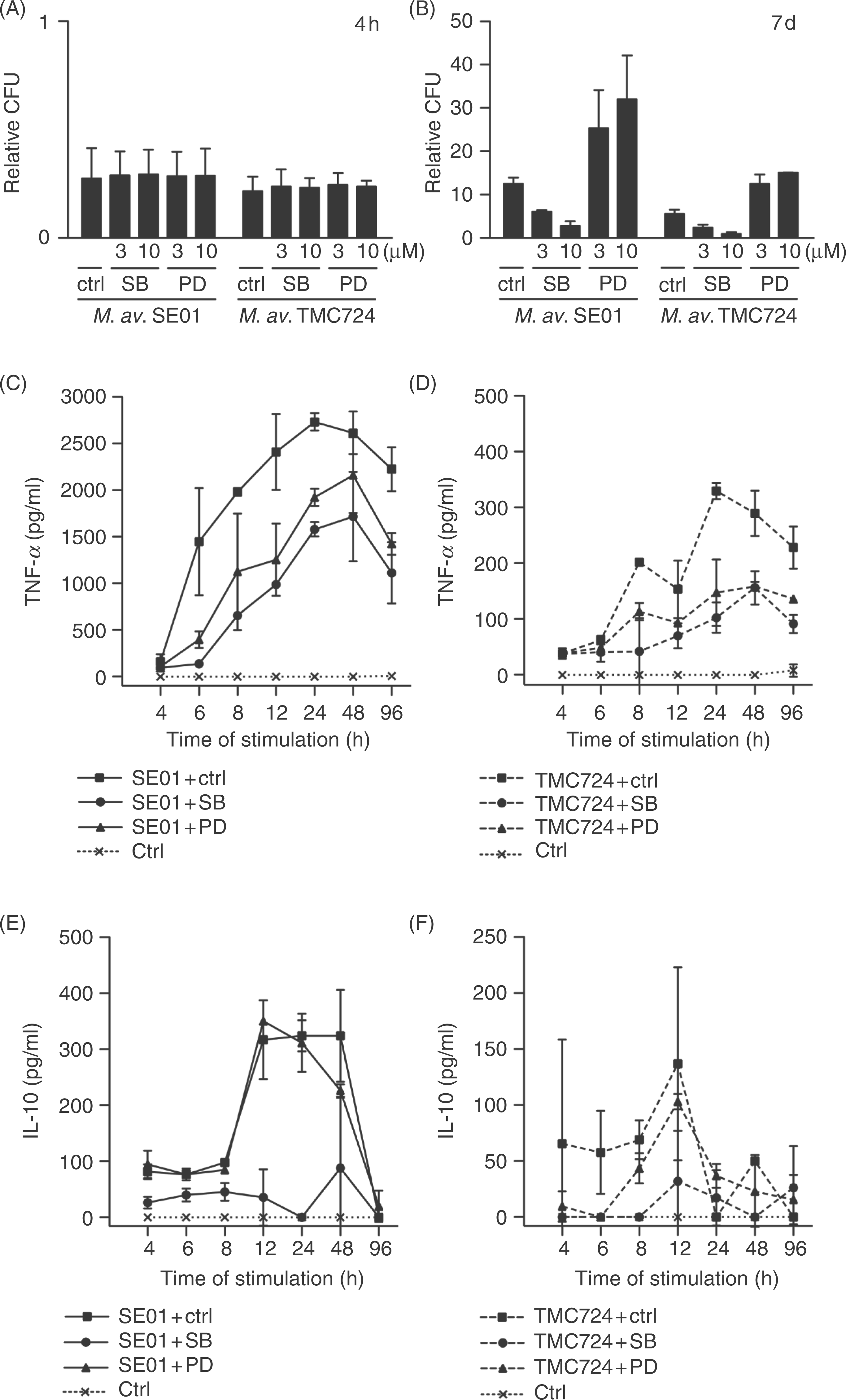
In contrast, the analysis on day 7 showed a dose-dependent reduction of at least 50% in relative CFU in macrophages with both M. avium isolates in the presence of SB203580 (Fig. 2B). The opposite was observed with the same isolates in the presence of PD98059, where a strong, dose-dependent increase of at least 100% in relative CFU in macrophages was seen compared to DMSO treated cultures. The solvent DMSO had no effect on uptake and replication (data not shown).
These data indicate that inhibition of p38 MAPK actively contributes to the control of M. avium in murine macrophages, whereas inhibition of the MKK/ERK pathway interferes with the antimycobacterial response and favors mycobacterial replication.
SB203580 reduces TNF-α and inhibits almost completely IL-10 production, whereas PD98059 reduces TNF-α but not IL-10 production
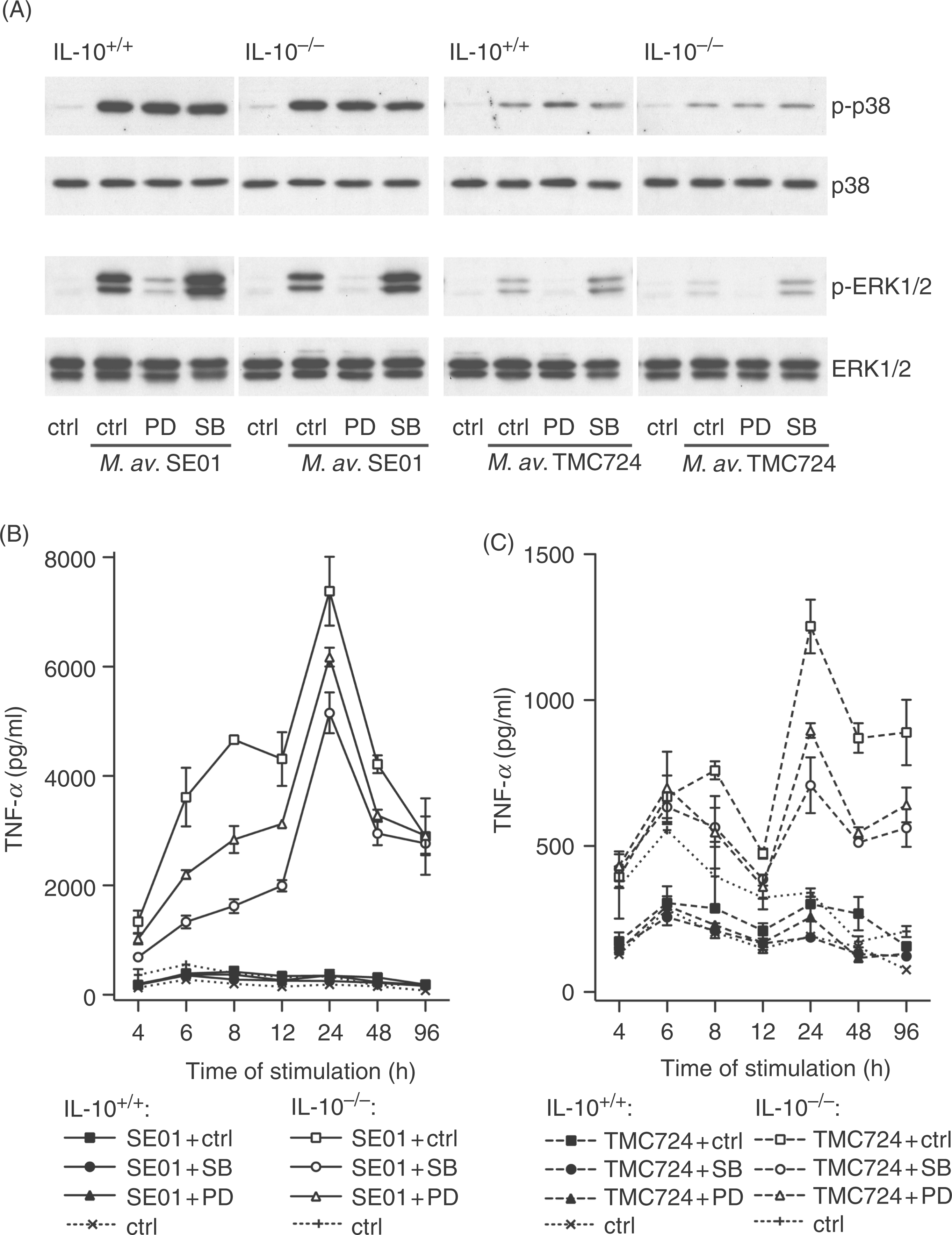
Macrophages were incubated with 10 µm of the MAPK inhibitors SB203580 or PD98059 and 0.1% (v/v) DMSO as solvent control for 1 h prior to and also during the infection with M. avium SE01 or TMC724. Mycobacterium avium-induced TNF-α formation of murine macrophages was reduced in the presence of PD98059 and even more in the presence of SB203580 (Fig. 2C,D). This was most prominently seen in experiments with M. avium SE01. After a rapid increase in the first 4–6 h of infection, a continuous increase in TNF-α levels was observed until 24 h, which was followed by a gradual decrease until 96 h.
The same samples were analysed for the presence of IL-10. The release of IL-10 by M. avium-infected macrophages was almost completely abolished over the whole detected time frame by treatment with SB203580, but remained unchanged in the presence of PD98059 (Fig. 2E,F). Compared to M. avium SE01-infected macrophages, M. avium TMC724-induced IL-10 production stayed near the lower limit of detection. Compared to TNF-α, the IL-10 production of M. avium-infected macrophages showed a different kinetic profile: maximum levels of IL-10 were detected from 12–48 h p.i., followed by a decline until 96 h. In solvent control treated macrophage cultures, no TNF-α and no IL-10 formation were observed during the experiment.
These data indicate that, in response to M. avium infection, SB203580 reduces TNF-α production and almost completely inhibits the IL-10 production, whereas PD98059 reduces the production of TNF-α but not IL-10.
The effect of SB203580 and PD98059 on M. avium replication in macrophages is independent of endogenously produced TNF-α
To address whether replication of M. avium is directly influenced via ERK1/2 and p38 or indirectly by the modulation of released cytokines, we analysed M. avium replication in TNF-α gene deficient mice. Tumor necrosis factor (TNF)-α−/− and TNF-α+/+ macrophages were incubated with 10 µm SB203580 or PD98059 for 1 h prior to, and also during, the infection with M. avium SE01 and TMC724. The uptake of both strains was similar in TNF-α+/+ and TNF-α−/− macrophages (data not shown). The replication rate of M. avium SE01 and TMC724 in TNF-α−/− macrophages compared to TNF-α+/+ macrophages was similar (Fig. 3). The TNF-α formation of both cell types was compared to ensure the functional lack of TNF-α formation in TNF-α−/− mice. Stimulation with M. avium as well as with LPS led to the expected TNF-α levels produced by TNF-α+/+ macrophages, whereas the cytokine was below detection limit in supernatants of TNF-α−/− cells (data not shown).
The influence of TNF-α on M. avium infection in murine macrophages and its modulation by SB203580 and PD98059. Tumor necrosis factor-α−/− and TNF-α+/+ macrophages were pre-incubated for 1 h with 10 µm SB203580 or PD98059 before additional incubation with 3 mycobacteria per macrophage and 50 ng/ml TNF-α. Cells treated with 0.1% (v/v) DMSO (inhibitor solvent) served as control (ctrl). Relative CFU determined 7 d p.i. in the absence or presence of exogenous added TNF-α. Shown are mean ± SD of three (TNF-α+/+ without mTNF-α: five) independent experiments. Data were analysed by multifactorial mixed model analysis (fixed factors: macrophage phenotype, bacterial strain, inhibitor, addition of cytokine) followed by Tukey–Kramer analysis where appropriate. P < 0.05 was considered significant; n.s., not significant.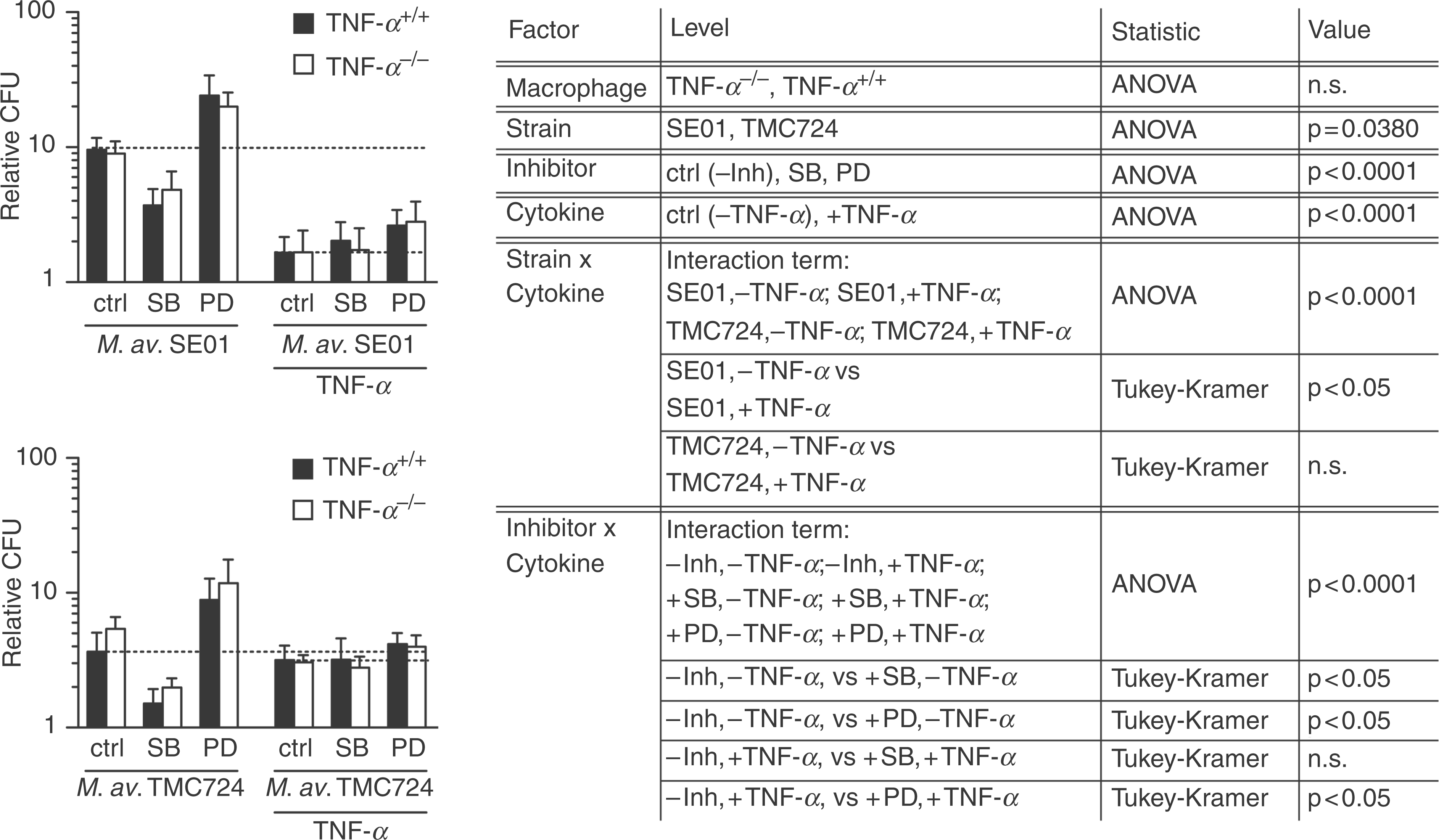
The presence of MAPK inhibitors strongly influenced M. avium replication in both, TNF-α−/− and TNF-α+/+ macrophages (inhibitor: P < 0.0001). Incubation with SB203580 resulted in a significant decrease (ctrl,–TNF-α vs +SB,–TNF-α: P < 0.05) of approximately 50% of relative CFU (Fig. 3). In sharp contrast, the presence of PD98059 led to a significant increase (ctrl,–TNF-α vs +PD,–TNF-α: P < 0.05) resulting in an increase of at least 120% of relative CFU compared to solvent control treated macrophage cultures without inhibitor (ctrl). These effects of SB203580 as well as PD98059 were observed in TNF-α−/− and TNF-α+/+ macrophages with M. avium SE01 and TMC724 in the absence of exogenously added TNF-α. Dimethylsulfoxide as solvent control of the used inhibitors had no effect (data not shown).
In parallel, we monitored whether the lack of endogenous TNF-α would influence the phosphorylation of MAPK in response to M. avium (Fig. 4A). We observed that signal intensities as well as the effect of the used inhibitors were comparable in TNF-α−/− and TNF-α+/+ macrophages: (i) the MAPK phosphorylation after infection with M. avium SE01 was enhanced compared to infections with TMC724; (ii) SB203580 did not influence p38 phosphorylation and caused stronger phosphorylation of ERK1/2; and (iii) incubation with PD98059 inhibited the phosphorylation of ERK1/2 but did not influence the phosphorylation of p38 MAPK. Unstimulated macrophages showed no phosphorylation of p38 and ERK1/2 in both cell types.
The influence of TNF-α on M. avium infection in murine macrophages and its modulation by SB203580 and PD98059. Tumor necrosis factor-α−/− and TNF-α+/+ macrophages were pre-incubated for 1 h with 10 µm SB203580 or PD98059 before additional incubation with mycobacteria. Cells treated with 0.1% (v/v) DMSO (inhibitor solvent) served as control (ctrl). (A) Phosphorylation of ERK1/2 and p38 MAPK during the M. avium infection. Macrophages were infected with M. avium SE01 and TMC724 at a MOI of 10 bacteria per macrophage and MAPK phosphorylation was analyzed at 30 min p.i. (details see Fig. 1). (B,C) Interleukin-10 formation in TNF-α−/− cultures infected with 3 bacteria per macrophage and analysed from 4 h to 96 h by ELISA, is shown as mean of duplicates ± SD of one representative trial out of two independent experiments (IL-10 formation in TNF-α+/+ cultures, see Fig. 2).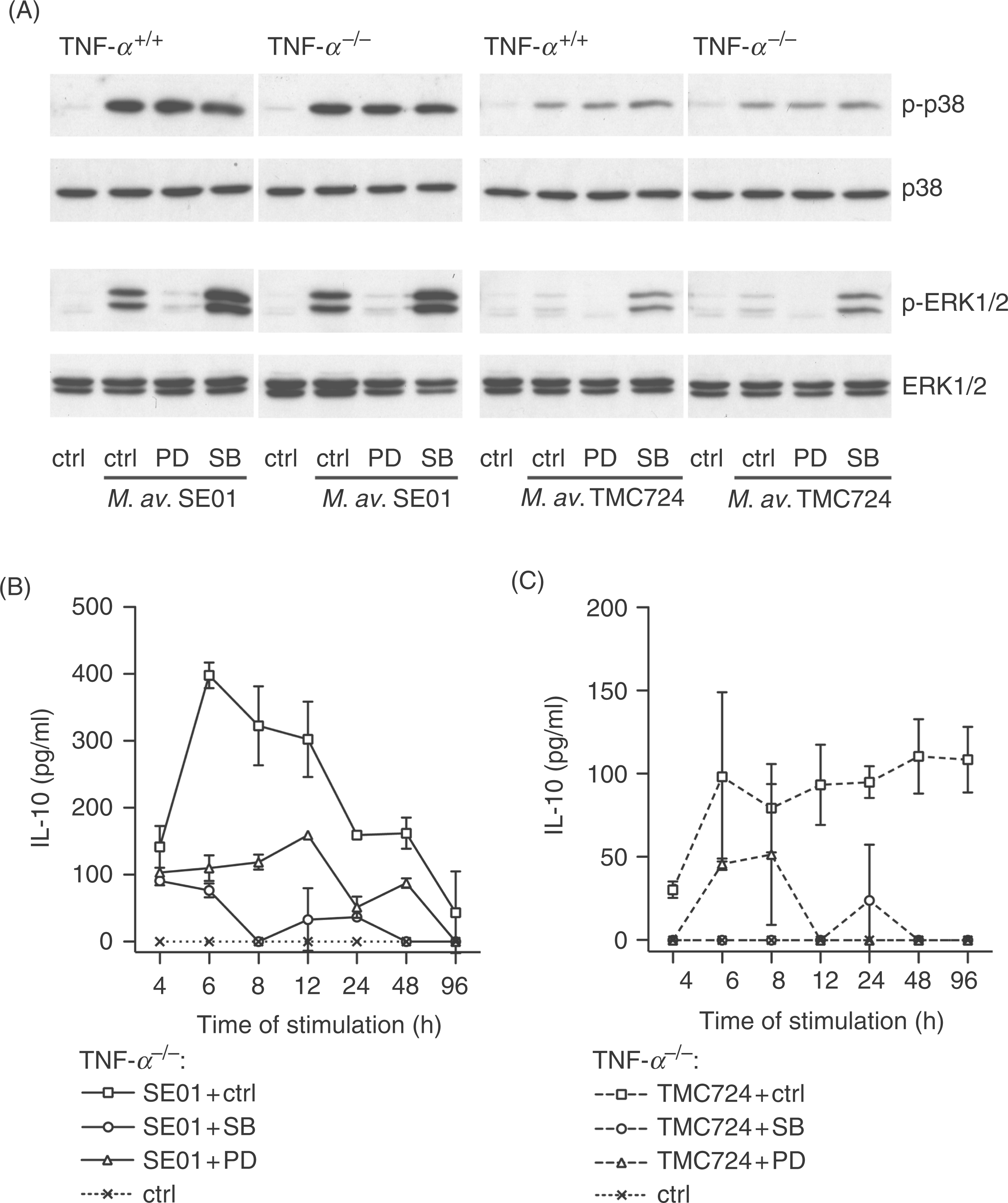
Taken together, these data indicate that endogenously produced TNF-α does not influence replication of M. avium regardless of the mycobacterial strain and the MAPK inhibitor used.
Exogenous, high-dose TNF-α over-rides the SB203580-mediated decrease of replication, and reduces the M. avium replication virulence-dependent
Since macrophage-derived endogenous TNF-α did not influence M. avium replication, we investigated whether excess amounts of TNF-α as expected in an inflamed tissue would show an effect in the presence or the absence of MAP kinase inhibitors. Tumor necrosis factor (TNF)-α−/− and TNF-α+/+ macrophages were incubated with 10 µm SB203580 or PD98059 for 1 h prior to and also during infection with M. avium SE01 or TMC724. At the time of infection, TNF-α was added at 50 ng/ml. This reduced the replication of M. avium SE01 significantly (+SE01,–TNF-α vs +SE01,+TNF-α: P < 0.05), but did not affect the replication of M. avium TMC724 (+TMC724,–TNF-α vs +TMC724,+TNF-α: not significant [n.s.]; Fig. 3). This strain-dependent effect of TNF-α was observed in both, TNF-α−/− and TNF-α+/+ macrophages.
The effects of the MAP kinase inhibitors were also affected by exogenously added TNF-α. The reducing effect of SB203580 on M. avium replication was over-ridden by exogenous TNF-α (ctrl,+TNF-α vs +SB,+TNF-α: n.s.), whereas the replication-promoting effect of PD98059 was still observed in the presence of exogenously added TNF-α (ctrl,+TNF-α vs +PD,+TNF-α: P < 0.05; Fig. 3). These effects were similar in M. avium SE01 and TMC724 infected TNF-α−/− and TNF-α+/+ macrophages.
Our data show that exogenously added TNF-α reduces the replication of M. avium SE01 but not of M. avium TMC724 and that it over-rides the replication-reducing effect of SB203580, but not the replication-promoting effect of PD98059 in M. avium infected macrophages.
The p38 MAPK exclusively regulates macrophage IL-10
We analyzed whether the results obtained with TNF-α−/− macrophages might be explained by altered IL-10 levels. So TNF-α−/− and TNF-α+/+ macrophages were incubated with 10 µm SB203580 or PD98059 or 0.1% (v/v) DMSO as control for 1 h prior to, and during, infection with M. avium SE01 or TMC724. The results for TNF-α−/− (Fig. 4B,C) compared to TNF-α+/+ (Fig. 2E,F) cultures infected with M. avium SE01 and TMC724 showed that the overall level of IL-10 production in both cell types was comparable. However, a difference in the kinetics was most prominently seen after infection of M. avium SE01. TNF-α−/− macrophages rapidly released IL-10 with a peak at 6 h p.i. and a rapid decrease thereafter, compared to TNF-α+/+ macrophages, which produced maximum levels of IL-10 on a plateau from 12–48 h p.i. and a decline until 96 h p.i. The use of SB203580 led to a nearly complete inhibition of the IL-10 production in both TNF-α−/− and TNF-α+/+ macrophages. The inhibitor PD98059 reduced the IL-10 production in TNF-α−/− macrophages. Dimethylsulfoxide, used as solvent control of the inhibitors, did not induce IL-10 release. In response to 50 ng/ml TNF-α, added at the time of infection, M. avium SE01 infected TNF-α−/− and TNF-α+/+ macrophages showed also an increased IL-10 production 48 h p.i., independent of the presence of PD98059 (data not shown). Again, SB203580 inhibited the IL-10 production almost completely. In contrast to SE01, TMC724 infected cells produced only little more IL-10 in the presence of 50 ng/ml TNF-α.
These data indicate that TNF-α influences the kinetics, but not the overall levels, of M. avium-induced IL-10 formation. In addition, the IL-10 formation of macrophages infected with M. avium in the presence and absence of endogenously and exogenously added TNF-α is exclusively dependent on p38 activation.
Lack of IL-10, but not exogenous IL-10, increases replication of M. avium
To segregate IL-10 mediated effects from those mediated by the p38 MAPK, the essential kinase in the signal transduction pathway of IL-10 production, we treated IL-10−/− and IL-10+/+ macrophages with the MAPK inhibitors and the inhibitor solvent control DMSO. Replication of M. avium SE01 and TMC724 was significantly higher in IL-10−/− than in IL-10+/+ macrophages (Fig. 5A), irrespective of the presence or absence of MAPK inhibition. We then monitored whether changes in the TNF-α formation of IL-10−/− macrophages would explain the enhanced M. avium replication in these cells. Compared to IL-10+/+ macrophages, the TNF-α release was about 10-fold higher in M. avium SE01 infected IL-10−/− macrophages and 2-fold higher in M. avium TMC724 infected IL-10−/− macrophages (Fig. 6B,C). In these cultures, TNF-α rose rapidly in the first hours with the first peak after 8 h and a very high second peak after 24 h followed by a moderate decrease until 96 h p.i. Additionally, we found in IL-10−/− and IL-10+/+ macrophages, SB203580 led to a stronger reduction of TNF-α compared to PD98059, independently of the M. avium strain used. Dimethylsulfoxide, used as solvent control of the inhibitors, did not induce TNF-α compared to solvent untreated cultures (data not shown). Of note, the addition of 10 ng/ml exogenous IL-10 to both cell types showed no effect on the replication of M. avium SE01 and TMC724 (Fig. 5B). In control experiments, we observed that the high TNF-α levels in IL-10−/− macrophages as well as normal TNF-α amounts in IL-10+/+ macrophages were reduced by the addition of 10 ng/ml IL-10 (data not shown), indicating that the amount of exogenously added IL-10 was biologically relevant.
The influence of IL-10 on M. avium infection in murine macrophages and its modulation by SB203580 and PD98059. Interleukin-10−/− and IL-10+/+ macrophages were pre-incubated for 1 h with 10 µm SB203580 or PD98059 before additional incubation with 3 mycobacteria per macrophage and 10 ng/ml IL-10. Cells treated with 0.1% (v/v) DMSO (inhibitor solvent) and 50 µm Tris-HCl (cytokine solvent) served as control (ctrl). (A,B) Relative CFU determined 7 d p.i. Shown are mean ± SD of three independent experiments. Data in (A) were analysed by three-factorial mixed model analysis (fixed factors: macrophage phenotype, bacterial strain, inhibitor); data in (B) were analysed by three-factorial mixed model analysis (fixed factors: macrophage phenotype, bacterial strain, addition of cytokine). Tukey–Kramer test was performed where appropriate. P < 0.05 was considered significant; n.s., not significant. The influence of IL-10 on M. avium infection in murine macrophages and its modulation by SB203580 and PD98059. Interleukin-10−/− and IL-10+/+ macrophages were pre-incubated for 1 h with 10 µm SB203580 or PD98059 before additional incubation with mycobacteria. Cells treated with 0.1% (v/v) DMSO (inhibitor solvent) served as control (ctrl). (A) Phosphorylation of ERK1/2 and p38 MAPK during the M. avium infection. Macrophages were infected with M. avium SE01 and TMC724 at a MOI of 10 bacteria per macrophage and MAPK phosphorylation was analyzed at 30 min p.i. (details see Fig. 1). (B,C) Tumor necrosis factor-α formation in IL-10−/− cultures infected with 3 bacteria per macrophage and analysed from 4 h to 96 h by ELISA, is shown as mean of duplicates ± SD of one representative trial out of three independent experiments.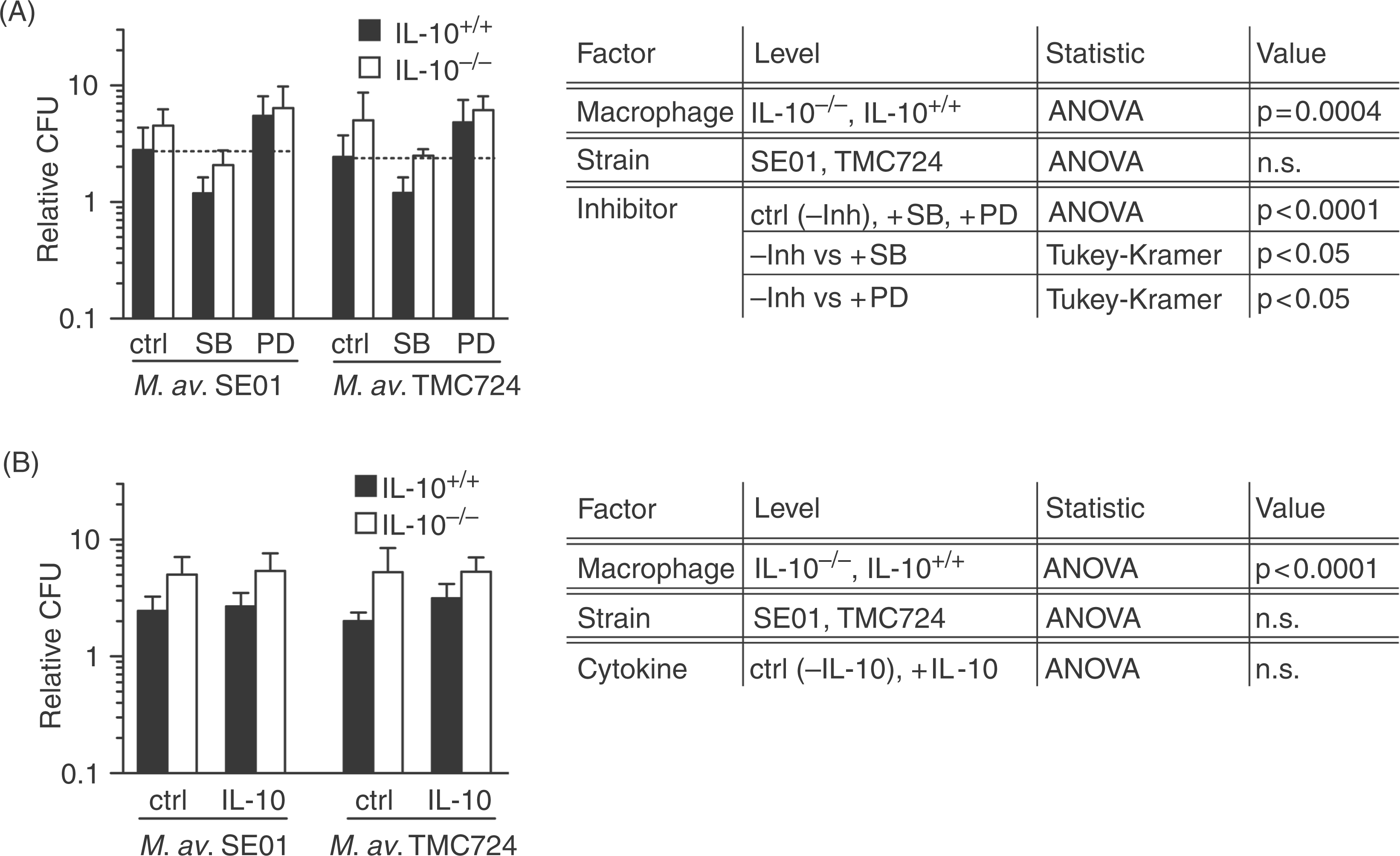
Interleukin-10 formation of both macrophage cell types was compared to ensure the functional lack of IL-10 formation in IL-10−/− mice. Stimulation with M. avium as well as with LPS led to the expected IL-10 levels produced by IL-10+/+ macrophages, whereas the cytokine was below detection limit in supernatants of IL-10−/− cells (data not shown).
These data indicate that in situations of low IL-10, but high TNF-α levels, M. avium replication is increased, which suggests that not a single cytokine but the TNF-α/IL-10 ratio in a given condition may determine the control of the mycobacterial growth.
The effect of SB203580 and PD98059 on M. avium replication in macrophages is independent of endogenously produced IL-10
Again, the effects of the MAPK inhibitors on replication were seen in IL-10−/− and IL-10+/+ macrophages infected with M. avium SE01 or TMC724 (inhibitor: P < 0.0001). Incubation with SB203580 resulted in significantly fewer relative CFU (ctrl vs +SB: P < 0.05), approximately 50% of solvent treated cultures (Fig. 5A). The replication in PD98059 treated macrophages was significantly increased by up to 100% (ctr. vs +PD: P < 0.05) compared to solvent control treated cells.
In parallel, we monitored whether the lack of endogenous IL-10 would influence the phosphorylation of MAPK in response to M. avium (Fig. 6A). We observed that signal intensities as well as the effect of the used inhibitors were comparable in IL-10−/−and IL-10+/+ macrophages: (i) the MAPK phosphorylation after infection with M. avium SE01 was higher compared to infections with TMC724; (ii) SB203580 did not influence p38 phosphorylation and caused stronger phosphorylation of ERK1/2; and (iii) incubation with PD98059 inhibited the phosphorylation of ERK1/2 but did not influence the phosphorylation of p38 MAPK. Unstimulated macrophages showed no phosphorylation of p38 and ERK1/2 in both cell types.
These results indicate that the p38 and ERK1/2 mediated effects on M. avium replication are independent of endogenous IL-10.
Discussion
Mitogen-activated protein kinases have previously been shown to critically influence mycobacterial replication.12,17,24 At least part of this effect was explicitly or implicitly attributed to the well-documented modulation of cytokines in infected macrophages by MAPK.16,21–
23
The current study is the first to show that p38 MAP kinase and the ERK pathway affect M. avium replication independently of endogenously produced TNF-α and IL-10 (Fig. 7).
The p38 MAPK and ERK1/2 pathway are critical regulators of M. avium replication in primary murine macrophages independent of TNF-α and IL-10. Graphic summary of infection experiments of M. avium (strains SE01 and TMC724) in wild-type (WT) and TNF-α- and IL-10-gene deficient macrophages (TNF-α−/−, IL-10−/−) using the MAPK inhibitors SB203580 (inhibited p38 pathway, open circle) and PD98059 (inhibited ERK1/2 pathway, filled circle), each at 10 µm, and as solvent control (ctrl, filled square) 0.1% DMSO (v/v). *P < 0.05.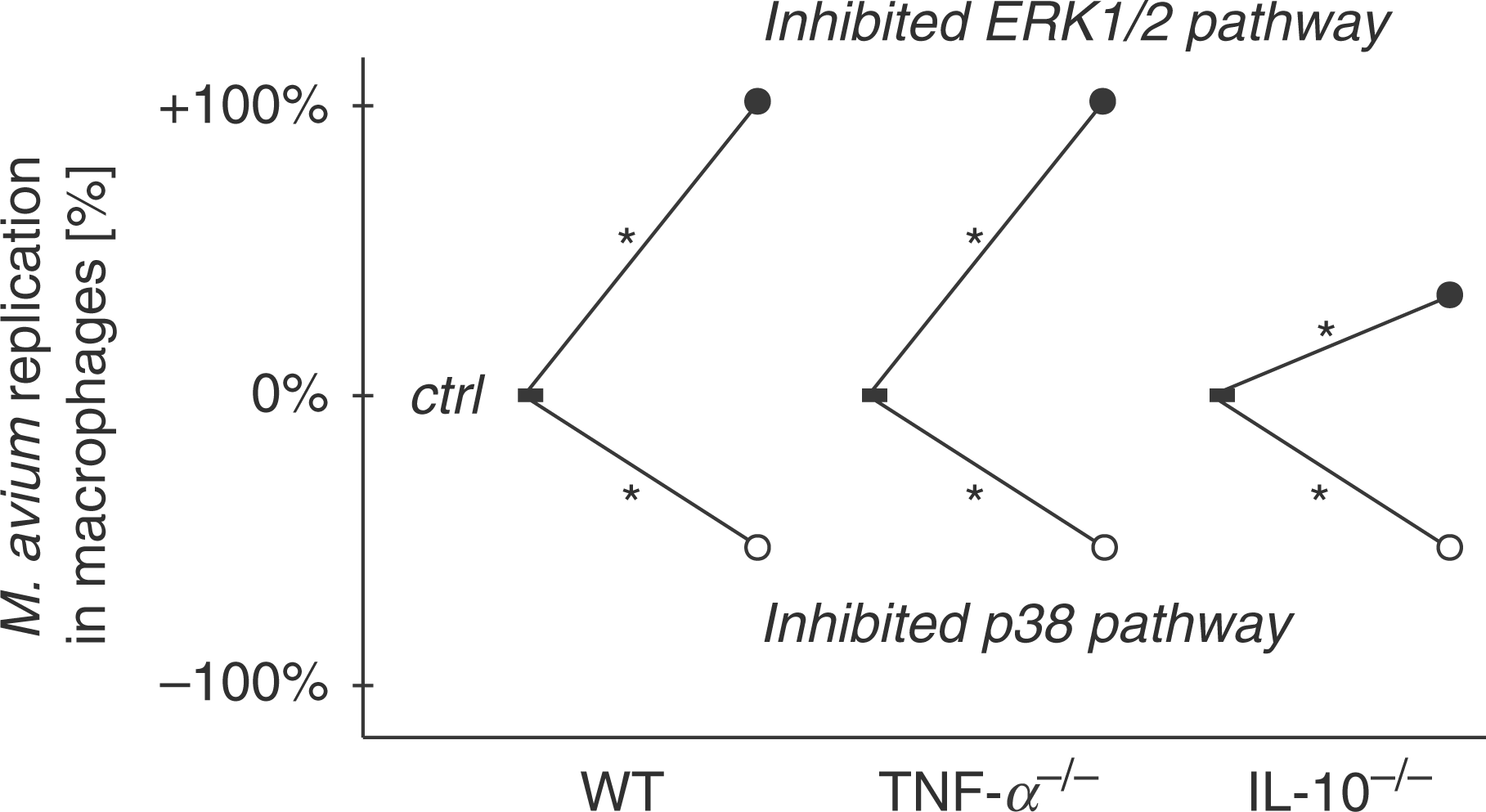
There is no clear picture in the literature as to whether MAPK inhibitors universally affect mycobacterial replication.12,17,24,26,27 Our own data in primary murine macrophages reinforce an emerging consensus that the replication of M. avium in murine macrophages is furthered by activated p38 and inhibited by activated ERK1/2. This can be asserted with some certainty because of the high degree of specificity of the inhibitors used. For example, SB203580 fails to affect the activities of a number of other protein kinases, including closely related MAPK family members. 19 The inhibitor SB203580 is a pyridinyl imidazole which inhibits specifically the activation of the two isoforms α and β of the MAPK p38 by binding to the ATP-binding site and inhibiting enzyme activity but not p38 phosphorylation.19,28
The inhibitor PD98059 is a well-established inhibitor of the ERK1/2 pathway. It was one of the first compounds to be described that targeted MKK1 and the closely related MKK2, and has been exploited in a multitude of subsequent studies. 29 PD98059 is a non-competitive inhibitor that appears to interact with the inactive unphosphorylated kinase more strongly than the active phosphorylated species and, therefore, exert their effects in cell-based assays by preventing the phosphorylation of MKK1, and/or the conformational transition that generates the activated enzyme.19,30
Treatment of macrophages with SB203580 during M. avium infection lowered bacterial replication rates. Which of the p38 MAPK mediated cellular processes favors mycobacterial replication is currently not known. Inhibition of mycobacteria-induced p38 MAPK activity caused a marked increase in EEA1 co-localization with mycobacterial phagosomes, and caused phagosomal acidification as well as enrichment of the late endocytic markers lysobisphosphatidic acid and CD63 (lysosomal integral membrane protein 1) on mycobacterial phagosomes. 31 Also, in bovine monocyte cultures, increased phagosome acidification, phagolysosome formation, and killing of mycobacteria was observed in the presence of SB203580. 27 Inhibition of p38 activity was shown to lead to increased inducible nitric oxide synthase (iNOS) protein levels in response to IL-1β. 32 It is possible that a similar mechanism is operative in mycobacteria-activated macrophages, enhancing the antimicrobial response.
The use of SB203580 in macrophages led to an enhanced phosphorylation of ERK as shown in Figures 4 and 6 and also by other investigators. 16 This observation may be explained by a potential inhibition of the PKB/Akt kinase, which has been reported at higher concentrations.19,33 Indeed inhibitors of AKT1 have been shown to counteract the bacterial manipulation of host signalling pathways, and were successfully used to limit the intracellular growth of M. tuberculosis and S. enterica sv. Typhimurium. 34 Since active PKB can act as a negative regulator of the activity of the ERK pathway, 35 inhibition of PKB may contribute to the enhanced activation of the ERK pathway, which suppresses mycobacterial growth. The latter is documented by this and previous studies in which inhibition of the ERK pathway led to increased mycobacterial replication in macrophages. The mechanisms by which ERK directly modulates macrophage function to limit mycobacterial replication is not clear to date. However, it has been shown that PD98059 inhibits nitric oxide formation in human astroglia. 36 In addition, PD98059 was reported to block the phosphorylation of p47(phox), 37 a key enzyme in the formation of reactive oxygen intermediates important for controlling bacterial replication. 38
In this context, a newly discussed model in which the p38 and ERK MAPK pathways tightly control autophagy also bears mentioning. It is proposed that p38 activity might limit the constitutive autophagy activity by reducing the maturation of autophagosomes. Conversely, autophagy stimuli such as starvation, that transiently activate ERK to a critical threshold might relieve this blockade and stimulate the maturation of autophagosomes. 39 This may, in part, provide an explanation of why ERK1/2 activity may be essential for the anti-mycobacterial armamentarium of murine macrophages, since it has been shown that autophagy is a critical effector mechanism to kill pathogenic mycobacteria. 40 Tumor necrosis factor-α is often thought to act in concert with other cytokines, such as IL-1 or IFN-γ, to activate macrophages for mycobacteriocidal activities. In line with this argument, the replication of M. avium strains 1983 and 2151 SmO was reported to be enhanced following treatment of murine macrophages with anti-TNF-α antibodies.11,17 However, the fact that replication of M. avium SE01 and TMC724 was similar in TNF-α−/− and TNF-α+/+ macrophages strongly suggests that endogenously produced TNF-α in an isolated in vitro system with primary macrophages does not play a major role in anti-mycobacterial mechanisms. This is corroborated by independent previous reports that neutralisation of TNF-α with anti-TNF-α antibodies did not affect the replication of the M. avium strains TMC724, 2151 SmO and SmT in murine bone marrow-derived and peritoneal macrophages.11,17,41
When the MAPK inhibitors SB203580 and PD98059 were used in infected TNF-α+/+ and TNF-α−/− macrophages, the results were indistinguishable. Therefore, in conditions where TNF-α levels are low or absent, the p38 MAPK and the ERK pathway dominate the regulation of mycobacterial replication in macrophages. In order to mimic a situation in which macrophages are in an environment of high TNF-α concentrations, we added 50 ng/ml TNF-α to the cultures. In these conditions, the inhibitory effect on the growth of the M. avium SE01 and TMC724 usually seen with SB203580 was no longer detectable (Fig. 8). Two explanations for this finding are possible. Tumor necrosis factor-α activates macrophages to a maximal extent that cannot be further enhanced by blocking p38, or alternatively, the p38 effect on M. avium growth reduction is antagonized, at least partially, by TNF-α. In contrast, the growth-enhancing effect of PD98059 was still evident when high TNF-α concentrations are present.
Addition of high exogenous TNF-α over-rides the SB203580-mediated inhibitory effects on M. avium replication in macrophages; however, it does not influence the PD98059-mediated increase in bacterial growth. Graphic summary of infection experiments of M. avium (strains SE01 and TMC724) in wild-type (WT) and TNF-α-gene deficient macrophages (TNF-α−/−) using exogenous TNF-α at 50 ng/ml, the MAPK inhibitors SB203580 (inhibited p38 pathway, open circle) and PD98059 (inhibited ERK1/2 pathway, filled circle), each at 10 µm, and as solvent control (ctrl, filled square) 0.1% DMSO (v/v). *P < 0.05; n.s., not significant.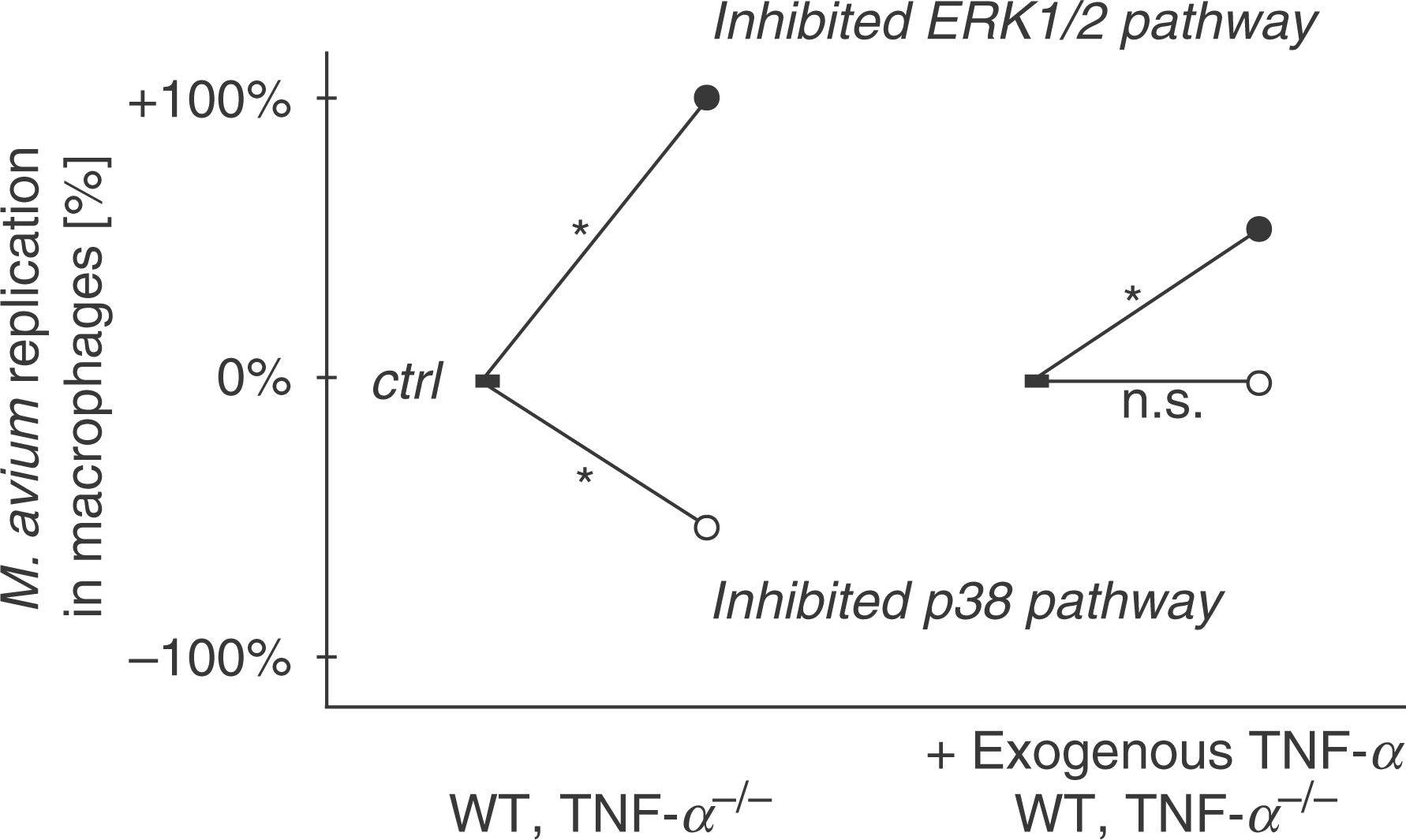
There is no doubt that TNF-α is essential for the development of protective immunity against M. tuberculosis and M. avium infection.42,43 This has been demonstrated in different experimental settings, including anti-TNF-α antibody treatment, TNFRp55- and TNF-α-deficiency. 44 Since endogenous macrophage TNF-α is apparently not involved in containing mycobacterial replication, the most relevant function of TNF-α in vivo lies in the recruitment of additional activated inflammatory cells to the site of infection, inducing granuloma formation and maintaining granuloma integrity.
Interleukin-10 is a potent anti-inflammatory cytokine which can modulate a number of cellular responses. 45 Compared with TNF-α, the regulatory role of IL-10 in mycobacterial infections is less clear, and conflicting results were reported concerning the replication of mycobacterial strains differing in their virulence in the absence of IL-10. While some laboratories showed that the absence of IL-10 did not significantly change the replication of mycobacterial strains differing in their virulence in vitro and in vivo,46,47 others found a decreased replication rate respectively a faster elimination of mycobacteria in vivo and in vitro using similar tools. 48 – 50
In our own experiments, M. avium SE01 and TMC724 showed an increased replication in IL-10−/− macrophages compared to IL-10+/+ macrophages. This is, at first sight, surprising, since very high TNF-α levels were evident in M. avium-infected IL-10−/− macrophages. Although the high TNF-α levels could be significantly reduced by exogenous IL-10, this did not affect the M. avium growth. Therefore, while IL-10 itself does not influence M. avium replication in murine macrophages, it appears that lack of IL-10 in the presence of high TNF-α levels actually favors M. avium replication in macrophages. A similar situation is found in cultures of TNF-α−/− and TNF-α+/+ macrophages following treatment with exogenous TNF-α and SB203580. The inhibitor almost completely inhibited IL-10 production, and, due to the addition of TNF-α, high TNF-α levels were present. In these cultures, we no longer observed the inhibitory effect of SB203580 on M. avium growth. Based on these data, we speculate that in conditions of very low to absent IL-10 (block by SB203580 or IL-10−/− macrophages), high TNF-α levels no longer reduce but promote M. avium replication in murine macrophages. This observation is reminiscent of the finding that, in human macrophages expressing low levels of IL-10, the addition of TNF-α actively promotes replication of virulent M. tuberculosis and Mycobacterium bovis. 51 The molecular mechanisms, which are involved in the TNF-α-induced increase in mycobacterial replication when the local IL-10 concentration is very low or even absent, are currently not known. Our data indicate that a certain amount of IL-10 is essential for the macrophage response but only if TNF-α levels are very high to mediate TNF-α-induced antimicrobial effector functions. Interleukin-10 has also been shown to affect the differentiation status of human myeloid cells. 52 Interleukin-10-treated dendritic cells turn into more macrophage-like cells, which show an enhanced anti-mycobacterial activity. It is possible that the lack of IL-10 in our experimental system reduced the anti-mycobacterial effector functions, which may explain the effects observed. However, detailed studies are needed to define whether these effects apply also to murine macrophages.
The regulatory influence of MAP kinase activity on mycobacterial replication has so far been mostly discussed to be indirect. The MAP kinase-dependent release of cytokines by macrophages was assumed to influence the outcome of an infection. The current study now clearly demonstrates that in isolated macrophages the effect of endogenously produced cytokines is by far not as important as previously thought. At the same time, this study also shows that the local cytokine concentration (as mimicked by the exogenous addition of high TNF-α), and the TNF-α/IL-10 ratio, is an important determinant of infection outcome in vitro, but likely under in vivo conditions as well.
Obviously, despite a very strong activation of the host cell response, the intermediately virulent M. avium SE01 is able to persist in macrophages. We have previously shown that M. avium SE01 induces up-regulation of the suppressor of cytokine signalling 1 (SOCS-1/SSI-1) and down-regulation of the IFN-γ receptor 1. 53 Both mechanisms effectively counteract the eradication of mycobacteria. We also identified M. avium SE01 to induce high IL-10 gene transcription. Taken together, M. avium SE01 is capable of tipping the scale of macrophage activation toward a net ‘anti-inflammatory’ score which favors persistence.
In our hands, both highly virulent and intermediately virulent M. avium strains are controlled by MAP kinase inhibitors. This demonstrates that a proper balance of MAP kinase activity is essential for the macrophage to control mycobacterial growth. The uniform response of the strains to the inhibitors may open a novel therapeutic avenue to treat mycobacterial infections, e.g. by selectively inhibiting p38 activity. However, our experiments with M. avium demonstrate that the efficacy of SB20350 to reduce mycobacterial replication depends on the local cytokine milieu. Inhibition of p38 MAPK activity may be of advantage in non-inflamed tissues with low concentrations of TNF-α, but our data imply that in the presence of high TNF-α concentrations the anti-mycobacterial effects would be substantially reduced. In contrast, our results point to a previously unappreciated opportunity for modulating mycobacterial replication, namely by stimulating ERK1/2 or activating ERK1/2-dependent mechanisms in infected macrophages. Therefore, the identification of genes specifically activated by ERK1/2 that are involved in mycobacteriostasis may lead to a more selective and targeted therapeutic approach that does not incur major inflammatory side effects.
Footnotes
Acknowledgements
The authors gratefully acknowledge the expert technical assistance of A. Hölscher, J. Volz, S. Kröger, S. Pfau and M. Richter at the Research Center Borstel. We thank Prof. T. Laskay and Prof. Dr W. Solbach for the opportunity to perform some cell isolation experiments at the Institute for Medical Microbiology and Hygiene at the University of Lübeck.
Funding
This study was funded in part by the Deutsche Forschungsgemeinschaft (DFG SFB415, Project C7 to NR und SE stimmt and the Cluster of Excellence ‘Inflammation at interfaces’, EXC306).