
Abstract
An as-yet limited body of evidence suggests that calcium-regulating endocrine hormones—in particular, parathyroid hormone–related peptide (PTHrP)—may have unappreciated cardioprotective effects. The current review focuses on the concept that PTHrP may, via modulation of classic cardioprotective signaling pathways, provide a novel strategy to attenuate myocardial ischemia–reperfusion injury.
Keywords
Acute myocardial infarction (AMI) remains one of the leading causes of death and disability worldwide: in-hospital mortality averages 8% to 15%, the estimated prehospital case fatality rate is 32%, and among survivors, the incidence of subsequent heart failure within 5 years postinfarction ranges from 6% to 32%. 1 -3 The current standard of care for AMI is timely reperfusion, thereby limiting the temporal progression of ischemia-induced cardiomyocyte death and reducing myocardial infarct size. However, despite its unquestionable benefits, restoration of blood flow to ischemic myocardium is, in itself, paradoxically associated with an element of cardiomyocyte death that may be responsible for as much as 50% of the final infarct size—a phenomenon termed lethal reperfusion injury. 4,5
Substantial time and resources have been devoted to the dual goals of obtaining insight into the molecular mechanisms of lethal reperfusion injury and identifying potentially promising strategies to mitigate cardiomyocyte death associated with reintroduction of blood flow. 5 -7 In this regard, there is growing interest in the molecular cross talk between the endocrine and cardiovascular systems, including the effects of hormones on cardiomyocyte viability in models of ischemic and oxidative stress. Although attention to date has largely focused on metabolic and reproductive hormones, 8 -14 there is nascent evidence that calcium-regulating hormones—in particular, parathyroid hormone–related peptide (PTHrP)—may have unappreciated cardioprotective effects. Our objective in this brief review is to explore the concept that PTHrP may, via modulation of classic cardioprotective signaling pathways, represent a novel approach to address a long-standing unmet therapeutic need, that is, reduction in myocardial infarct size, beyond that achieved by reperfusion alone. 6,7,15
The Problem: Ischemia–Reperfusion Injury
The immediate therapeutic goal in the setting of AMI is to reestablish patency of the infarct-related artery in a timely manner, thereby restoring blood flow to the at-risk myocardium and limiting myocardial infarct size. 15 However, while reintroduction of oxygen and nutrients is essential for the salvage of ischemic cardiomyocytes, reperfusion has been described as a “double-edged sword,” that is, can rescue one population of myocytes while precipitating the death of others. 16 This paradox has been termed lethal reperfusion injury—or, to more accurately reflect the continuum of the process, lethal ischemia–reperfusion injury. 4,5,15,17
The molecular mechanisms responsible for ischemia–reperfusion-induced cardiomyocyte death are complex and multifactorial (reviewed in 4,17,18 ). Nonetheless, 4 decades of investigation have identified 3 pivotal and integrated events: generation of cytotoxic reactive oxygen species (ROS) upon reintroduction of oxygen to the ischemic tissue, cytosolic and mitochondrial calcium overload (a consequence of normalization of ischemia-induced acidosis, influx of sodium via sodium–hydrogen exchange, and subsequent sodium–calcium exchange) and, ultimately, destabilization of mitochondria culminating in opening of the mitochondrial permeability transition pore (mPTP), collapse of mitochondrial membrane potential, and uncoupling of oxidative phosphorylation. 4,5,17 -24 Efforts have been made to mitigate lethal ischemia–reperfusion injury via administration of pharmacologic agents targeting each of these harbingers of cardiomyocyte death. Treatment with ROS scavengers, calcium channel antagonists, and inhibitors of mPTP opening all showed promise in many (but not all) preclinical studies; however, subsequent evaluation in clinical trials was disappointing. Indeed, none of these strategies have been successfully translated from preclinical models into clinical practice. 4,5,7,15,25,26
The Gold Standard for Cardioprotection: Ischemic Conditioning
The search for therapeutic strategies to attenuate lethal ischemia–reperfusion injury was transformed by landmark observations that myocardial infarct size can be favorably influenced by (1) exposing the at-risk region of the heart to brief, nonlethal episodes of antecedent ischemia before the onset of the prolonged ischemic insult 27 ; (2) initiating reperfusion in a stuttered or staccato (rather than abrupt) manner 28,29 ; or interestingly (3) applying brief periods of transient ischemia–reperfusion in a remote tissue or organ, before, during, or upon relief of sustained myocardial ischemia. 30 -33 Each of these 3 “conditioning” paradigms—ischemic preconditioning, postconditioning, and remote conditioning—have, with few exceptions, been shown to evoke a profound infarct-sparing effect in all experimental models studied (reviewed in 18,34 -37 ). Thus, not surprisingly, the effects of ischemic conditioning (most notably, postconditioning and remote conditioning, which are more clinically applicable as prophylaxis is not required) on surrogate serum biomarkers of infarct size and longer-term end points encompassing morbidity and mortality are under investigation in phase 2 and phase 3 trials. 18,36 -46
Common Mechanistic Themes: Role of the Reperfusion Injury Salvage Kinase Pathway
Preconditioning, postconditioning, and remote conditioning are distinguished by inherent differences in the timing and site at which the cardioprotective stimulus is applied. Nonetheless, despite these intrinsic differences (and despite the added complexity of remote conditioning in which the cardioprotective trigger must be transferred or communicated from the distant tissue to the heart 34,47,48 ), all 3 conditioning paradigms appear to share common mechanistic themes. Specifically, at the level of the cardiomyocyte, there is an overall agreement that ischemic conditioning is initiated by ligand–receptor binding (ie, release of autacoid, endocrine, and/or paracrine signaling molecules and subsequent stimulation of G-protein-coupled receptors on the sarcolemmal membrane) and propagated via activation of one or more intracellular signal transduction pathways. 18,34,37,49 -54 The intracellular signaling pathways evoke favorable modifications in one or more subcellular end effectors (with proposed candidates including, but not limited to, mitochondria, the sarcoplasmic reticulum, and other regulators of ionic homeostasis)—modifications that culminate in rendering the cardiomyocyte less vulnerable to lethal ischemia–reperfusion injury. 18,34,37,49 -54
The postreceptor signal first implicated to play a role in ischemic preconditioning is the production of the second messenger 1,2-diacylglycerol (DAG: achieved via receptor-mediated activation of phospholipase C and/or D and subsequent hydrolysis of phosphatidylcholine or phosphatidylinositol 4,5-bisphosphate) and the resultant activation/translocation of protein kinase C (PKC). 55 -57 Ensuing studies refined this seminal “PKC hypothesis” (most notably, elucidated the specific contribution of the ∊-isoform of the kinase) 58 and expanded the paradigm to investigate the potential involvement of a host of signaling pathways (in concert with or as alternatives to PKC signaling) in the spectrum of ischemic preconditioning, postconditioning, and remote conditioning. In this regard, although multiple signaling cascades have been proposed to play a role, 18,34,37,49 -54,59,60 recruitment of the so-called “RISK” (Reperfusion Injury Salvage Kinase) pathway, involving phosphorylation of extracellular signal-regulated kinase (ERK)1/2 and/or phosphatidylinositol 3 (PI3) kinase/Akt, has been identified in many (but not all) models as necessary and sufficient for conditioning-induced cardioprotection. 60 -65
A Balancing Act: Kinases and Phosphatases
Regulation of the RISK pathway—and, indeed, regulation of all kinase signaling—is achieved by the orchestrated balance between kinase activation (typically achieved by phosphorylation) and dephosphorylation. 66 Of particular relevance in conditioning-induced cardioprotection is the potential involvement of mitogen-activated protein kinase phosphatase 1 (MKP-1), a phosphatase that contributes to the dephosphorylation of members of the mitogen-activated protein kinase (MAPK) family including p38 MAPK, c-Jun N-terminal kinase, and, notably, ERK1/2. 67,68 Mitogen-activated protein kinase phosphatase 1 has been shown to play a regulatory role in multiple and diverse pathophysiologic processes, including (but not limited to) innate and adaptive immunity, metabolic homeostasis, stem cell–mediated skeletal muscle regeneration, and maintenance of bone mass. 68 Given our current understanding of the mechanisms of ischemic conditioning, logic would suggest that MKP-1-mediated control of kinase signaling may, similarly, be of importance in conditioning-induced cardioprotection. Although the overwhelming majority of studies published to date have focused exclusively on the kinase phosphorylation component of the kinase–phosphatase equation, this concept is supported by evidence for a loss in the infarct-sparing effect of postconditioning in murine models of aging and diabetes displaying attenuated ERK1/2 phosphorylation and an accompanying increase in the expression of MKP-1. 69,70 The consequences of modifying the balance between ERK1/2 phosphorylation and dephosphorylation in heart will, in all likelihood, be complex and model dependent. 71 Nonetheless, the limited evidence available to date is consistent with a role of MKP-1-mediated regulation of ERK phosphorylation in the mechanism of cardioprotection.
Parathyroid Hormone–Related Peptide
Parathyroid hormone–related peptide was first discovered in the 1940s in the setting of hypercalcemia in patients with cancer and further identified in the 1980s as a paraneoplastic protein with the ability to activate parathyroid hormone (PTH) receptors. 72 As the name suggests, there is a homology between PTHrP and PTH, that is, 16% of the genetic sequence (1-14 N-terminal amino acid) and structure of PTHrP resembles that of PTH, thereby allowing it to bind to and activate type 1 parathyroid hormone receptor (PTH1R).
Despite this structural similarity, PTHrP is not primarily secreted from the parathyroid gland; rather, the expression and secretion of PTHrP is complex and multifaceted. It is well known that, under pathological conditions, PTHrP is secreted from certain types of tumor cells leading to humoral hypercalcemia of malignancy. In addition, there is a growing appreciation that PTHrP is a polyhormone that plays a physiologic role in intracrine, autocrine, and local paracrine signaling in multiple tissues. Examples include (but are not limited to) (1) the skeleton, where PTHrP coordinates the rate of chondrocyte differentiation to maintain the orderly growth of long bones during development 73 ; (2) the mammary glands, where PTHrP signaling is required for the formation of breast tissue 74 ; (3) the placenta, where PTHrP is thought to promote calcium transport from mother to fetus and be required for the maintenance of normal fetal calcium concentration 75 ; (4) the pancreatic islets, where PTHrP has been linked to regulation of β-cell mass, insulin production, and inhibition of apoptosis 76,77 ; and (5) smooth muscle (including cardiovascular tissue), where it is secreted in response to mechanical deformation and functions in a short feedback manner to relax stretched muscle. 78 -80
Classic PTHrP Signaling
The classic, well-established effects of PTHrP, elucidated largely in bone, are mediated via binding to PTH1R (a G-protein-coupled receptor) and activation of both adenylyl cyclase/protein kinase A pathway and phospholipase C signaling (Figure 1). 81 Activation of the adenylyl cyclase pathway results in the transcription of PTH target genes, a process regulated by protein kinase A–induced phosphorylation of transcription factors such as cyclic adenosine monophosphate (cAMP) response element-binding protein. Activation of the phospholipase C arm of the intracellular signaling pathway results in the generation of DAG (as described previously for ischemic conditioning) and, in parallel, the production of inositol 1,4,5-trisphosphate (IP3), a second messenger generated by the hydrolysis of phosphatidylinositol 4,5-bisphosphate and, interestingly, also implicated to play a role in the infarct-sparing effect of ischemic preconditioning. 82,83 Increased intracellular concentrations of IP3 initiate the release of calcium from the endoplasmic reticulum while, as in the setting of ischemic conditioning, DAG is the substrate for the activation and subcellular translocation of PKC. The identity of the specific PKC isoform(s) activated by PTHrP–PTH1R–phospholipase C-DAG signaling has not been resolved. However, there is evidence to suggest that PTHrP-mediated mitogenesis of pancreatic β cells is associated with the expression of PKC-ζ, 77 while PTH1R stimulation by PTH has been reported to activate multiple members of the PKC family (including the α and β isoforms) in bone cells. 84,85 Despite these aforementioned gaps in knowledge, both components of the phospholipase C pathway have been demonstrated to regulate the expression of transcription factors in the nucleus (Figure 1). 86

Classic PTHrP signaling. Classic PTHrP signaling is initiated by binding of the hormone to PTH1R (a G-protein-coupled receptor) and activation of both the AC/PKA and PLC pathways. Activation of AC triggers the transcription of PTH target genes, a process regulated by PKA-induced phosphorylation of transcription factors such as CREB. Activation of PLC results in the generation of both DAG and IP3 (second messenger generated by the hydrolysis of PIP2) and, via subsequent activation and subcellular translocation of 1 or more isoforms of PKC, serves to regulate the expression of transcription factors in the nucleus. PLC indicates phospholipase C; AC, adenylyl cyclase; PIP2, phosphatidylinositol 4,5-bisphosphate; IP3, inositol 1,4,5-trisphosphate; DAG, diacylglycerol; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; PKC, protein kinase C; PKA, protein kinase A; PTHrP, parathyroid hormone–related peptide; PTH1R, type 1 parathyroid hormone receptor.
Parathyroid Hormone–Related Peptide and Regulation of MAPKs
More recently, a third downstream signaling pathway triggered by PTH–PTHrP–PTH1R binding has been identified: PTH and PTHrP have been demonstrated to regulate the activity of MAPKs. 87 -91 The effects of PTH/PTHrP on MAPKs are complex, may be cell specific, and, in some instances, appear to be discordant. For example, PTH1R stimulation reportedly initiates cAMP-mediated activation of MAPK in Chinese hamster ovary R15 cells 87 and inhibits MAPK in an osteosarcoma cell line, 88 while in osteoblasts, both activation of MAPKs (in particular, ERK1/2) in proliferating cells and inhibition of MAPKs in differentiated cells have been described. 89 -92 Although the molecular mechanisms responsible for this complex regulation of ERK1/2 activity with PTH1R signaling remain incompletely understood, evidence obtained in bone has revealed that PTH/PTHrP-mediated alterations in phosphatase expression—specifically, expression of MKP-1—play a pivotal role. 91
Parathyroid Hormone–Related Peptide and Mitochondrial Stability
As discussed previously, destabilization of mitochondria is considered among the major molecular mechanisms contributing to ischemia–reperfusion-induced cardiomyocyte death. Limited evidence obtained from noncardiac cell culture models subjected to proapoptotic stimuli (ie, chemotherapeutic agents) suggest that PTHrP may attenuate mitochondrial-dependent apoptosis and favor the preservation of mitochondrial integrity via modulation of casein kinase 2 activity, expression, and trafficking 93 and induction of antiapoptotic B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma-extra large (Bcl-xl) in concert with downregulation of proapoptotic Bax protein. 94 In contrast, in a naive embryonic cell line, stimulation of the PTH–PTHrP–PTH1R axis reportedly upregulates (rather than inhibits) mitochondrial-dependent apoptosis, 95 leading to speculation that the effect of PTH1R activation on mitochondrial integrity is “bidirectional” and determined by cell type and the maturation or differentiation state of the cell. 95 Whether this concept of a “bidirectional effect” of PTHrP on mitochondrial-dependent apoptosis can be extrapolated to cardiomyocytes—thereby implying that PTHrP may contribute to mitochondrial stabilization in ischemic-reperfused myocardium—remains to be established. In addition, the possible role of PTHrP-mediated suppression of MKP-1 in the antiapoptotic (or proapoptotic) effect of the peptide and the effect of PTHrP-PTH1R signaling on the stability of the mPTP are at present unknown.
Cardiovascular Effects of PTHrP: Is PTHrP a “Conditioning Mimetic?”
There is evidence that the requisite components of PTHrP-PTH1R signaling are present in the heart and vasculature, 80,96 -99 that PTHrP functions as a mechanosensitive regulatory molecule and participates in the control of vascular tone (and thus blood pressure), chronotropy, and inotropy, 80,96,100 -102 and that PTHrP may also play an as-yet poorly defined role in a spectrum of cardiovascular pathologies. In support of this latter concept, release of PTHrP and activation of PTHrP-PTH1R have been reported in the settings of heart failure, ischemia/hypoxia, and ischemia–reperfusion in both preclinical models and humans. 96,97,103 This information, together with our understanding of the molecular mechanisms of PTHrP–PTH1R signaling and the common mechanistic themes shared with PTHrP–PTH1R signaling and ischemic conditioning, raises the question: is PTHrP cardioprotective? More precisely: does PTHrP act as a conditioning mimetic and limit infarct size via inhibition of MKP-1 and subsequent upregulation of survival kinase signaling? (Figure 2).
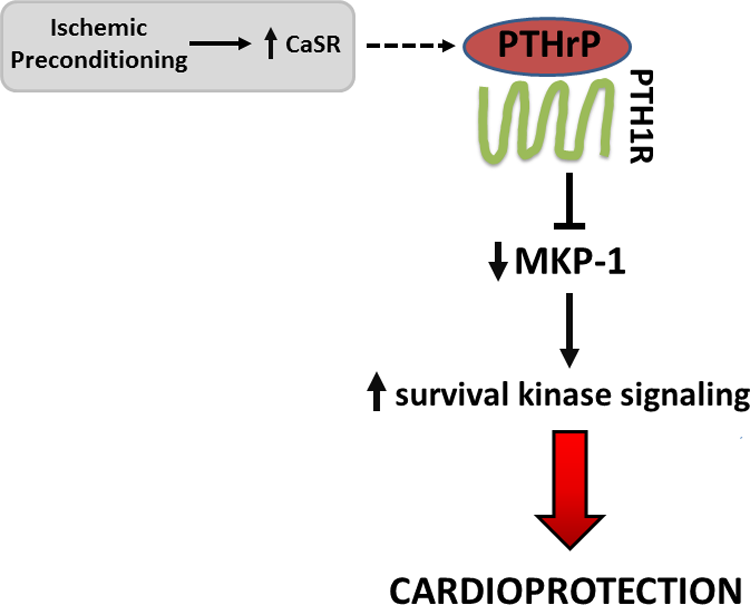
Parathyroid hormone–related peptide signaling and cardioprotection. Parathyroid hormone–related peptide, administered exogenously or secreted in response to endogenous triggers (ie, ischemic preconditioning and subsequent activation of the extracellular CaSR), may render cardiomyocytes resistant to lethal ischemia–reperfusion injury via inhibition of MKP-1 and upregulation of survival kinase signaling. PTHrP indicates parathyroid hormone–related peptide; CaSR, calcium-sensing receptor; MKP-1, mitogen-activated protein kinase phosphatase 1.
Among the small number of published studies in which PTHrP has been investigated in models of ischemia–reperfusion or hypoxia–reoxygenation, there is an overall agreement that direct administration of PTHrP, or increased cardiac expression of PTHrP-PTH1R as a secondary consequence of thyroxine-induced hyperthyroidism, confers a protective effect. 104 -107 However, the primary end point in these studies was not infarct size (the gold standard of cardioprotection); rather, these studies focused on recovery of contractile function following relief of ischemia or other stressors. 104 -106 Interestingly, recovery of contractile function with PTHrP treatment was augmented in females when compared to males, a finding that is consistent with the observed role of estrogen in modulating PTHrP expression. 105 One additional study, in which infarct size was among the primary end points, provides indirect support for the concept that PTHrP may act as a conditioning mimetic. Specifically, activation of the extracellular calcium-sensing receptor in heart—the primary physiologic function of which is to regulate the local secretion of PTHrP—was shown to contribute to the infarct-sparing effect of ischemic preconditioning via mechanisms involving upregulation of ERK1/2 phosphorylation and RISK signaling. 108
Finally, and most recently, a direct link between exogenous administration of PTHrP, RISK signaling, and cardiomyocyte fate has been established. 109 In isolated adult murine cardiomyocytes subjected to ROS-induced stress (as discussed previously, a pivotal component of lethal ischemia–reperfusion injury), viability was significantly improved in cells pretreated with PTHrP versus untreated controls (75% vs 25%, respectively; Figure 3). The favorable effects of PTHrP treatment were accompanied by a downregulation in the expression of MKP-1 and were abrogated by pharmacologic inhibitors of MAPKs and PI3 kinase/Akt. Further corroboration was provided in experiments utilizing cardiomyocytes harvested from MKP-1 null mice: knockout of MKP-1 had the predicted effect of rendering cardiomyocytes resistant to ROS-induced death, with no significant benefit provided by administration of PTHrP. 109

Parathyroid hormone–related peptide protects cardiomyocytes from H2O2-induced oxidative stress. A, Pretreatment with PTHrP attenuated H2O2-induced cell death of primary cardiomyocytes. Representative images of cardiomyocytes isolated from adult male 29J/C57BL6 wild-type mice showing viable (rod-shaped) and dead (round-shaped) cells in response to 5 to 20 minutes of 100 µM H2O2 exposure in the presence or absence of 100 nM PTHrP. B, Quantitative analyses of viable cells after 10 to 15 minutes of oxidative stress are shown. Viable cells were calculated and averaged as a percentage of total number of cells in 3 to 4 different random fields. The results were expressed as the means of 4 to 5 independent experiments and plotted. a P < .001. Reprinted with permission from Datta et al.109 PTHrP indicates parathyroid hormone–related peptide.
Administration of PTHrP: A Tenable Therapeutic Approach
Before administration of PTHrP can be considered a tenable therapeutic strategy for the treatment of myocardial ischemia–reperfusion injury, 2 fundamental questions must be addressed. The first and obvious gap in our current knowledge is: does PHTrP reduce infarct size? Although the aforementioned studies provide implicit evidence, the concept that PTHrP may have an infarct-sparing effect has, to date, not undergone direct and robust in vivo evaluation in any preclinical studies in which the reduction in infarct size is the primary end point.
A second and equally important issue for potential clinical application is: can an infarct-sparing effect of PTHrP be safely achieved in the absence of confounding side effects? Although the maximum, safe dose of PTHrP has not been identified, one study conducted in humans has reported that, in the setting of osteoporosis, doses of PTHrP ranging from 500 to 625 µg/d, administered subcutaneously for 3 weeks, were not associated with adverse effects, while doses greater than 750 µg/d resulted in mild hypercalcemia. 110 No significant effects on arterial pressure or heart rate were observed at doses of 500 to 1500 µg/d, 110,111 with the exception of an episode of postural hypotension in one individual. 110
Importantly, if PTHrP were to be administered as an infarct-sparing strategy in the setting of AMI, short-term, intravenous (IV), or intracoronary infusion of the agent at the time of reperfusion (rather than chronic subcutaneous injection) would presumably be a more a relevant paradigm. In this regard, the consequences of sequential IV infusion of escalating concentrations of PTHrP, at rates of 0.9 to 14.9 µmol/min (or approximately 0.03-0.5 µg/min) for 15 minutes, has been investigated in healthy human volunteers. Infusion of the peptide did not alter total or ionized serum calcium and had no effect on arterial pressure but, at higher concentrations, was associated with tachycardia. 112 Initial data obtained in conscious rats suggested that PTHrP may also have a direct, positive inotropic effect. 113 However, more recent studies revealed that PTHrP has no direct effect on contractile elements, 114 and no inotropic consequences were observed with IV infusion of PTHrP in healthy humans. 112 Accordingly, in order to ascertain whether administration of PTHrP is a feasible therapeutic strategy, evidence will be required demonstrating a reduction in infarct size under conditions in which (1) PTHrP is given IV or via local intracoronary infusion, (2) treatment is initiated at the onset of reperfusion, and (3) the effective dose is not confounded by tachycardia.
Future Directions
It would be premature, based on the limited amount of available data, to propose that PTHrP is a “better” and more promising candidate than the multiple agents that have been evaluated previously. Nonetheless, the apparent action of PHTrP on multiple components of cardioprotective signaling, and the novelty of MKP-1 as an as-yet unexplored target, may potentially confer an advantage. Taken together, our current understanding of lethal ischemia–reperfusion injury, ischemic conditioning, and PTHrP-PTH1R signaling raises the intriguing possibility that PTHrP may represent a novel strategy to address the longstanding unmet need of reducing infarct size beyond that achieved by reperfusion alone.
Footnotes
Author Contributions
Tanuka Datta and Karin Przyklenk contributed to acquisition and interpretation and drafted the manuscript. Nabanita S. Datta was involved in interpretation. All authors are involved in conception, manuscript revision, and final approval of the manuscript and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.