
Abstract
Background:
Fluid therapy is universally administered in the management of patients with sepsis, however excessive cumulative fluid balance has been shown to result in worse outcomes. Hyperoncotic albumin results in both lower fluid volumes and early cumulative fluid balance, and may reduce short-term mortality in patients with septic shock.
Methods:
In this single centre, open label, feasibility trial; patients with early septic shock will be randomly allocated either 20% albumin for resuscitation and daily supplementation, versus buffered crystalloids alone for all fluid therapy. The intervention period will last 7 days, with follow up points at ICU and hospital discharge, and 90 days after randomisation.
Objectives:
Primary outcome measures including recruitment rate, intervention adherence, data completeness and safety will constitute objective evidence of feasibility, according to pre-specified thresholds. Secondary outcomes will include mortality and healthcare utilisation at 90 days, alongside other physiological and patient centred outcomes to inform the design of a future effectiveness trial.
Conclusion:
This study will rigorously test the feasibility of conducting a future trial to test both the clinical and cost-effectiveness of hyperoncotic albumin in patients with early septic shock.
Introduction
Sepsis represents an increasing burden on healthcare systems, with over 19 million cases per year globally, resulting in approximately 5 million deaths. 1 International clinical practice guidelines for the management of sepsis recommend doctors urgently give intravenous fluid to patients, however this strong recommendation is based on low quality evidence. 2 There are also data linking the administration of excessive volumes of fluid with worse outcomes for patients with sepsis.3 –5 In addition, there is very little evidence to guide clinicians in the choice of fluid to use. Salt solutions, known as crystalloids, are recommended in the first instance, but human albumin solution is suggested for patients with persistent evidence of hypoperfusion after receiving a large volume of fluid.
Hyperoncotic albumin preparations are presented in lower (50–100 ml) volumes compared to their isotonic counterparts. In previous work, we have shown their use results in lower fluid resuscitation requirements and is associated with lower early cumulative fluid balance in critically ill patients.6,7 Other investigators have utilised hyperoncotic albumin as a regular supplement. Dubois et al. found regular supplementation with 200 ml 20% albumin per day resulted in reduced measures of organ dysfunction and lower fluid balance compared with controls. 8 In the ALBIOS trial, patients with sepsis were randomised to receive daily supplementation with 20% albumin titrated to daily serum measurements, versus crystalloid fluid alone. 9 Mortality did not differ significantly between groups, however in the sub-group of patients with septic shock at enrolment (n = 1121), there was a lower risk of death for those receiving 20% albumin, which remained after adjustment for baseline covariates.
We have recently conducted a systematic review summarising all available evidence for hyperoncotic albumin in patients with sepsis. There is weak evidence suggesting a short-term mortality benefit in patients with septic shock, but very little evidence to support its broader use in patients with sepsis. 10 The review also found hyperoncotic albumin to be safe and associated with a lower short-term cumulative fluid balance and faster resolution of shock, yet better quality trials are required to increase the certainty of this evidence. This trial is designed to test the concept of hyperoncotic albumin, both as a resuscitation fluid and regular daily supplement in adult patients with early septic shock.
Study objectives
Primary objective
To establish whether it is feasible to identify and randomise patients with early septic shock to receive 20% albumin for fluid resuscitation and daily supplementation versus crystalloid therapy alone, in UK critical care units according to this study protocol.
Secondary objectives
To collect pilot data on clinically important outcomes in the intervention and comparator groups to inform the design of a future effectiveness study.
To pilot data collection tools through virtual follow-up and health economic questionnaires.
To embed streams of experimental medicine to better understand the potential mechanistic effects of hyperoncotic albumin in patients with septic shock.
Methods and analysis
Trial design and setting
An open-label, single-centre, randomised (1:1) parallel group, feasibility trial with allocation concealment (see Figure 1). This study will be conducted at the intensive care units of two NHS university teaching hospitals, Manchester Royal Infirmary and Wythenshawe Hospital. These hospitals operate under Manchester University NHS Foundation Trust, comprising a total of 57 critical care beds.
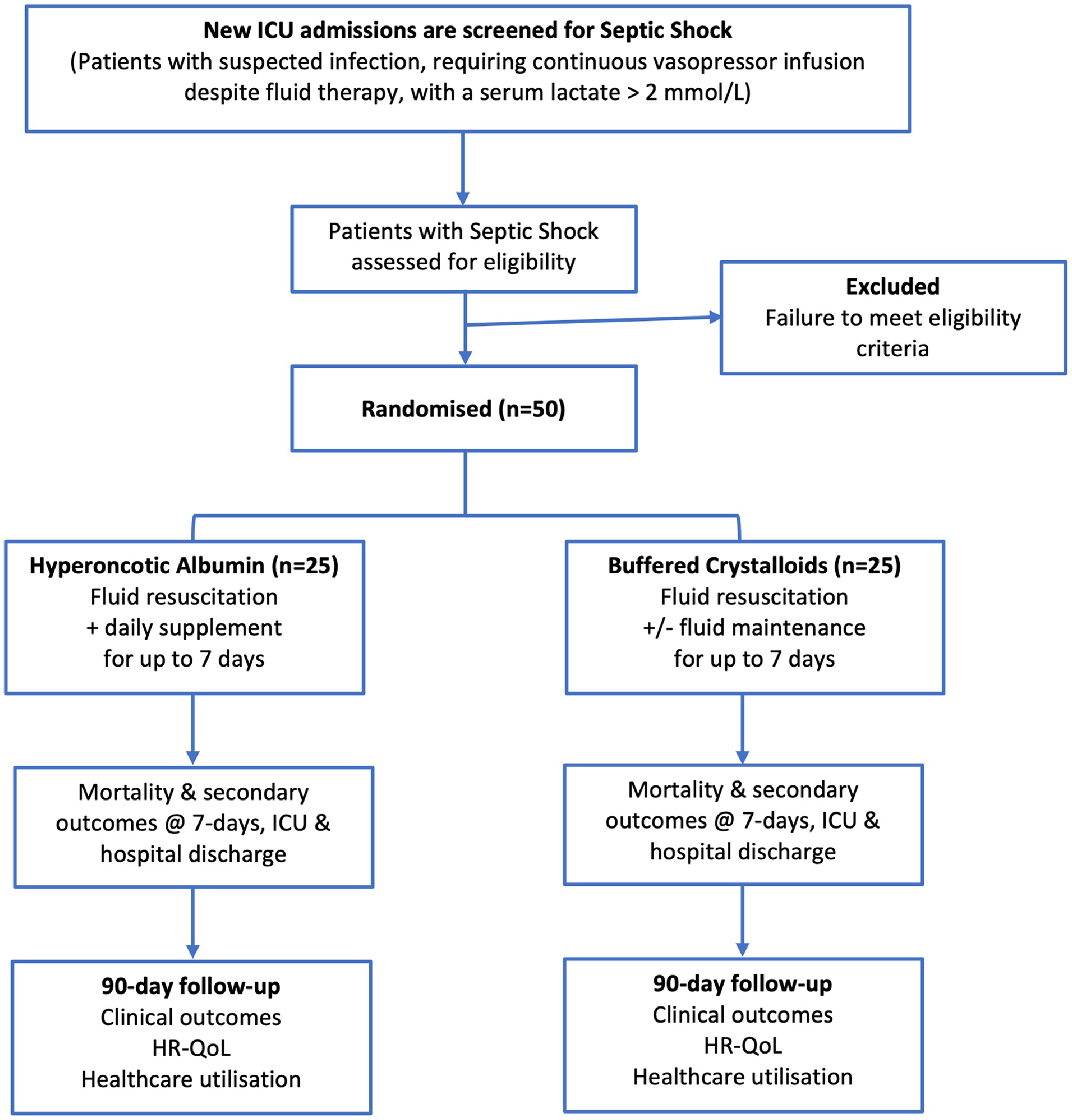
Study schematic diagram.
Patient & public involvement
Members of the Manchester University Critical Care User Group have been directly involved in the design and planning of this study. We elicited patient and public views through structured engagement events on the conduct of this study. We have gained their focussed views concerning the study’s consent procedures and the acceptability of sampling and storage of blood samples.
Patients who have suffered septic shock and others who experienced prolonged ICU stays have agreed to continue to contribute towards the design and delivery of this study, including involvement in the trial steering committee. Their involvement will serve to ensure this study and any future trials remain considerate of the needs of participants and their families.
Participants & interventions
Screening & eligibility criteria
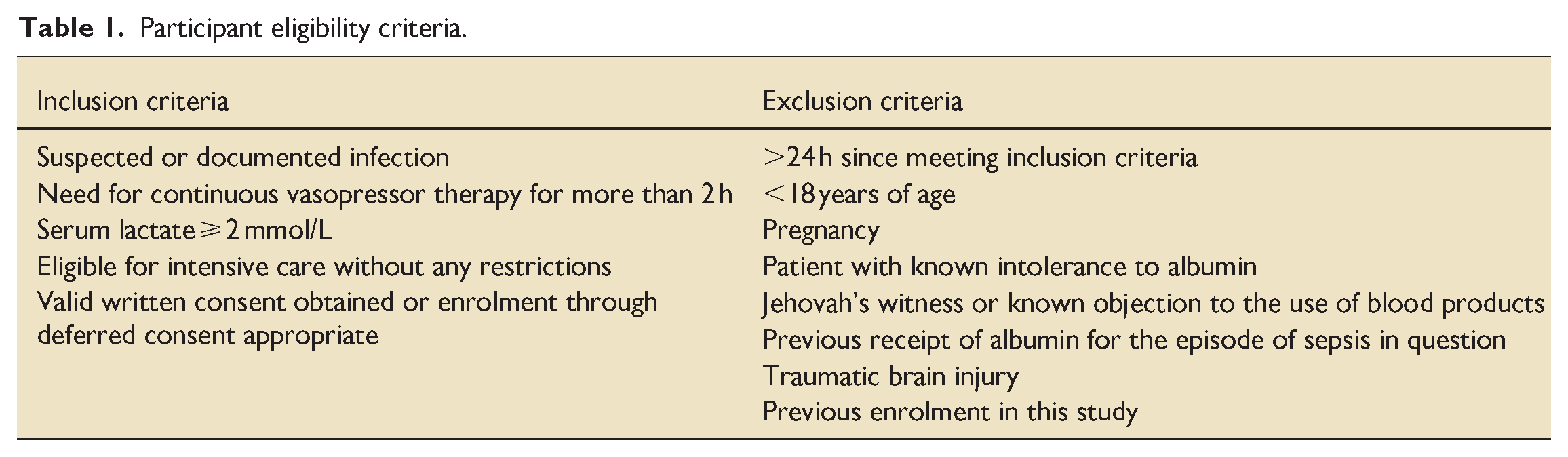
New ICU admissions will be screened for eligibility by trained and delegated members of the research team. Eligibility will be confirmed by a medically qualified person who is experienced in the assessment and management of patients with septic shock. Eligibility criteria are detailed in Table 1.
Participant eligibility criteria.
Intervention description
Participants in the intervention group will receive 20% albumin for all intravenous fluid resuscitation at a dose and rate determined by the treating physician. They will also be administered daily intravenous supplementation of 20% albumin for up to 7 days. Dosing of albumin supplementation will be guided by daily serum albumin measurements (see below). The use of crystalloid solutions for the purposes of fluid therapy will be discouraged. As patients in both groups will receive crystalloid fluids as dilutants for drug infusions, the use of crystalloids in the intervention arm will not be considered a protocol deviation.
Dose adjustment according to daily serum albumin concentration:
⩾30 g/l: no albumin required ⩾25 and <30 g/l: 20 g over 1–2 h twice daily (40 g total daily dose) ⩾20 and <25 g/l: 20 g over 1–2 h three times daily (60 g total daily dose) <20 g/l: 20 g over 1–2 h four times daily (80 g total daily dose)
The treating ICU physician will determine the volume, rate of administration and time-point of prescribing all resuscitation fluids (both intervention and comparator arms). Daily albumin supplements maybe prescribed by either a treating ICU physician or a medically qualified study investigator. All intravenous fluid, in both trial arms will be administered by the bedside nurse according to usual local established practice.
Intervention discontinuation
Twenty percent albumin will be administered for up to 7 days after randomisation. Recent analysis of data from over 200 consecutive patients admitted to our ICU with confirmed septic shock, revealed median [IQR] values for time dependent on vasopressors and ICU length of stay to be 5.1 [2.4–11.5] and 6.7 [3.1–13.0] days respectively. Therefore, the 7-day intervention period will encompass the majority of patients’ vasopressor requirements and time spent in critical care. The intervention will be discontinued at 7 days, or at discharge from critical care (whichever comes sooner), or when contraindication to albumin administration arises (see daily dosing schedule above).
Comparator description
Patients allocated to the comparator arm will receive buffered crystalloids for all fluid resuscitation and maintenance requirements. These solutions are already in widespread use at all participating ICUs. The dose and rate of administration will be determined by the treating physician. Physicians will be advised to follow the surviving sepsis guidelines for fluid resuscitation, which are already in widespread use at participating ICUs. 2 Any use of albumin for patients in the comparator arm will be reported as a protocol deviation.
Intervention adherence
Prior to opening study recruitment, investigators will provide comprehensive training for local clinical staff on this protocol and how to deliver the study intervention. Emphasis will be placed on prohibiting the use of albumin in the comparator arm and strongly discouraging use of crystalloids in the intervention arm.
The intervention will be prescribed and administered by the treating clinical team according to usual local practice. Members of the research team will supervise and support accurate and reliable delivery of the intervention daily. This will test the embedding of the intervention into clinical care pathways whilst ensuring sufficient oversight of adherence by local investigators and research staff.
Outcomes
Primary outcome: Feasibility
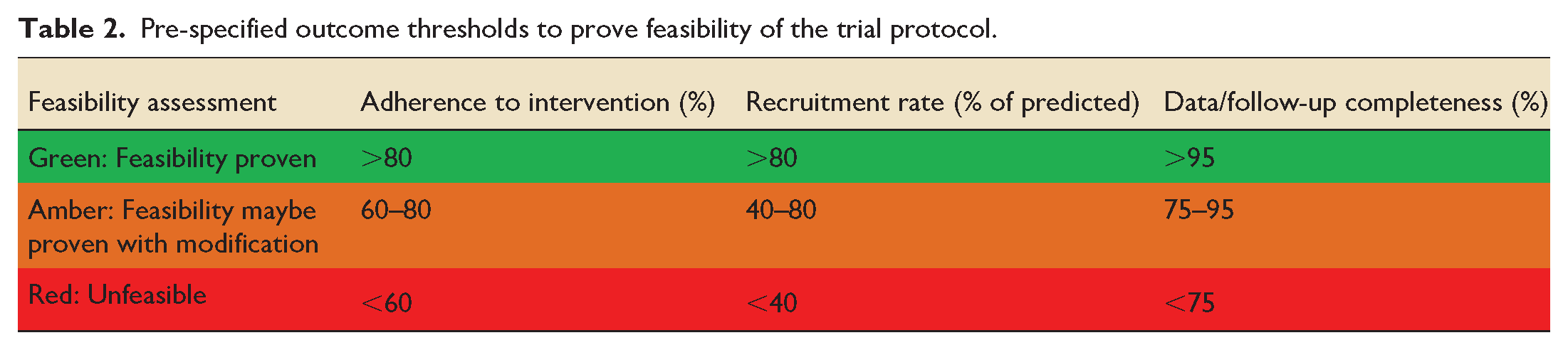
Evidence of feasibility in successful delivery of this study protocol is the primary objective of this trial. The objective measures of feasibility are described below and their thresholds shown in Table 2.
Adherence to the intervention: Adequate adherence to trial interventions will be defined as at least 80% of participants receiving the intervention they were allocated, during the first 7 days, without any cross-over between groups. Quantitative data on protocol deviations along with qualitative data on reasons for non-adherence will also be collected.
Recruitment rate: Success will be defined as achievement of at least 80% of the recruitment target without any major modification of the study protocol. If the recruitment rate is in the range from 40% to 80% of the target, this may support a future trial if the rate can be improved through modifications of the study protocol, use of additional sites or both. Support for a future definitive trial would be unlikely if recruitment is below 40% of the target within the given period.
Follow-up and data completeness: Success will be defined as 95% completeness of datasets and follow-up. Data completeness rates of 75%–95% may also represent success if >90% of data for the likely primary outcome for a future effectiveness trial have been captured; or if provision of additional site training on dataset completion is supported. If data completeness is below 75%, progression to a further trial would only be supported if there is a clear and realistic plan to improve compliance.
Safety: Data for adverse events will be collected for participants in both trial arms. All reported adverse events will be discussed at monthly TMG meetings and made available for scrutiny by the TSC. Any adverse event that resulted in halting of the trial on safety grounds would render proof of feasibility null and void.
Pre-specified outcome thresholds to prove feasibility of the trial protocol.

Secondary outcomes
Data on other outcome measures will further confirm feasibility and provide information on important clinical outcomes. The latter combined with our review findings will be used to inform the sample size calculation of a larger definitive randomised trial. We will also pilot methods for a future cost-effectiveness analysis.
Further measures of feasibility
Time from consent/randomisation to the first dose of study fluid
Randomised to screened patient ratio
Reasons for exclusion
Rates of consent refusal and participant drop-out
Costs of fluid therapies in each arm
Physiological outcomes (measured daily up to 7 days post randomisation)
Fluid input, output & balance
Hourly vasopressor dose requirements & mean arterial pressure
Indices of cardiac output (where available)
Serum lactate
Measures of organ dysfunction (e.g. SOFA score)
Patient-centred outcomes
Survival and free from vasopressors at day 7 post randomisation
Time spent in the ICU and hospital post randomisation
Mortality 90-days post randomisation
Incidence of new or worsening acute kidney injury (KDIGO classification) in the first 7 days post randomisation
Need for renal replacement therapy at 7- and 90-days post randomisation
Healthcare service use and costs at 90 days
Health related quality of life at baseline and day 90, measured using the EQ-5D-5L
Participant timeline
The schedule of events for a trial participant’s timeline is shown in Table 3.
Schedule of enrolment, interventions and assessments.
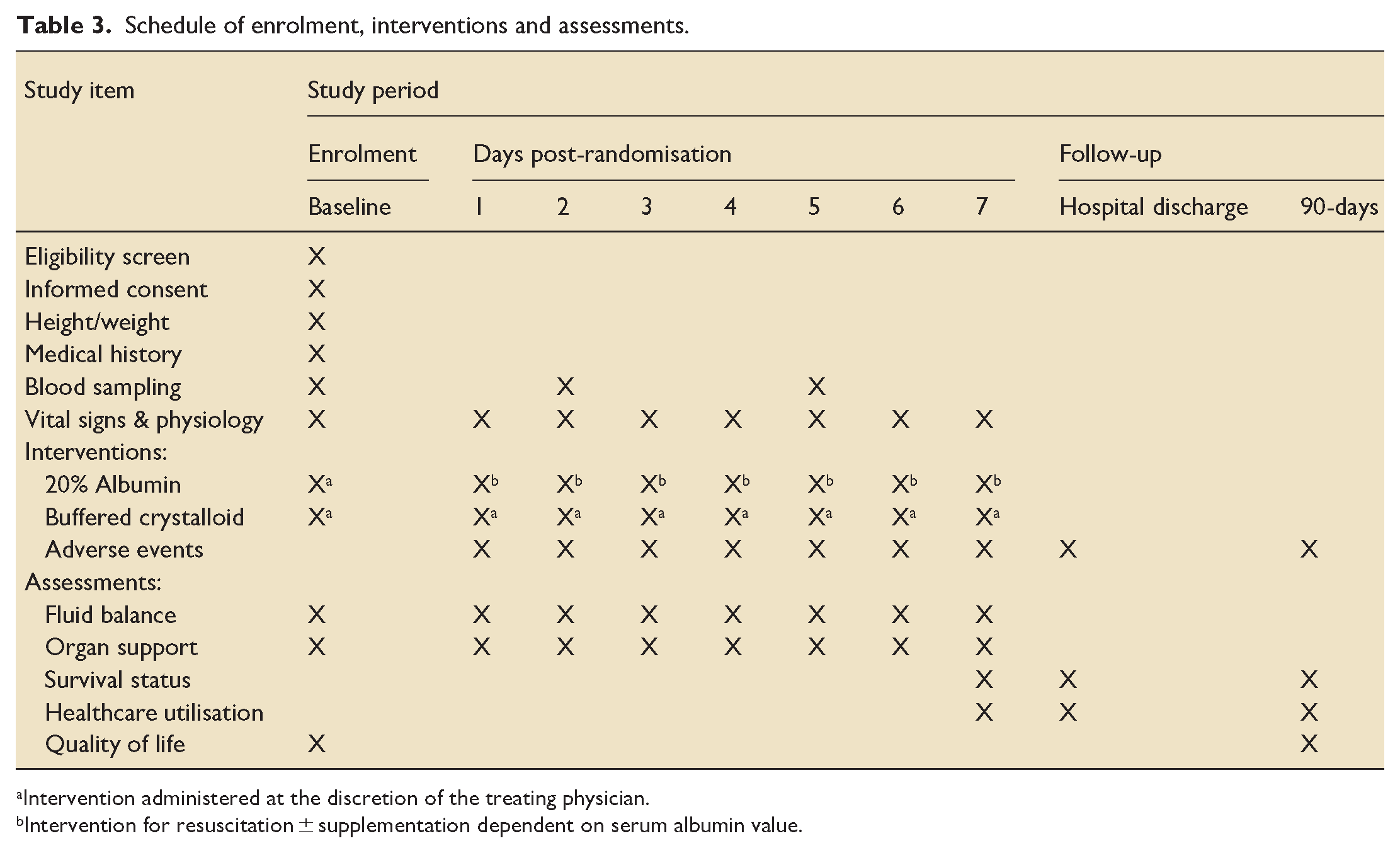
Intervention administered at the discretion of the treating physician.
Intervention for resuscitation ± supplementation dependent on serum albumin value.
Sample size
As this is a feasibility study, a formal power calculation is not appropriate. This trial is not powered to detect a clinically meaningful difference between the two treatment groups for any outcome. Accordingly, the target is to recruit 50 participants to provide adequate assessment of feasibility outcomes in relation to progression to a more definitive trial. This will enable us to detect an 80% success rate for recruitment with 95% binomial confidence intervals of 66.3%–90.0%.
Randomisation
Randomisation will be performed as soon as possible after confirming eligibility. Patients will be randomised by trained research staff, using the web-based software sealed envelope (www.sealedenvelope.com), following a central computer generated 1:1 sequence to either the intervention group (20% albumin) or comparator (buffered crystalloids) using this dedicated online randomisation service available 24 h/7 days per week. Central computer-generated randomisation should preserve allocation concealment, but as this is a small feasibility study, it is not possible to randomise in permuted blocks of varying size.
Blinding
As 20% albumin is provided in 100 ml glass bottles and buffered crystalloids in plastic bags, blinding of participants, clinical staff and members of the research team in this small feasibility trial will not be possible. Research staff conducting 90-day follow-up and the trial statistician will be blinded to treatment allocation, to minimise any potential bias affecting the assessments they make.
Data collection & analysis
Participating sites currently log all patient data into harmonised electronic medical records (Epic Systems; Verona, WI, USA). This includes regular automated import of physiological observations, organ support parameters and results from both laboratory and radiology departments. This existing infrastructure will ensure the highest quality of primary source data for many of our measured outcomes.
Follow-up outcomes will include healthcare utilisation (further hospital admissions until day-90) and health related quality of life (EQ-5D-5L). Blinded assessors independent of the trial team, will be trained on how to conduct such assessments reliably and robustly at 90-days via telephone. They will complete electronic case report forms, which will then be transcribed into the study database by an unblinded member of the research team.
Statistical analysis
All participants’ data collected will be collated and summarised for analysis. As this is a feasibility study, formal significance testing will not be performed. Appropriate descriptive statistics and graphics will be utilised to explore the distribution and completeness of all variables. Feasibility measures will be presented as percentages with 95% binomial confidence intervals. Descriptive statistics for all secondary outcome measures will be presented for baseline and follow-up time-points (days 1–7 & 90 post-randomisation). Differences between treatment groups with associated 95% confidence intervals will be estimated based on an intention to treat analysis. As all trial interventions are already in established clinical use, we will also perform a per protocol analysis and compare the results for patients who crossover between arms.
Economic analyses
Within this feasibility study, we will pilot methods for the collection of cost and health related quality of life (HRQoL) data for a future cost-utility analysis. Healthcare resource use from baseline until 90-days will be collected using patients’ medical records and a study-specific questionnaire. This will include the comparative costs of fluid therapies in each arm. These data will include the duration and level of critical care received, hospital length of stay and therapeutic procedures received during the primary admission and any readmissions. Trial-based cost-utility analyses (CUAs) require the measurement of HRQoL for the calculation of quality adjusted life years (QALYs). However, baseline measurement of HRQoL in the critically ill is problematic because patients are often incapacitated at the time of randomisation. In this study we will compare two approaches for obtaining a baseline/initial assessment of HRQoL using the EQ-5D-5L by:
Asking a family member (where available) to rate the patients HRQoL before they were admitted to critical care via the EQ-5D-5L and
Patient self-completion of the EQ-5D-5L at the earliest time possible post-randomisation (at the time of consent to continue).
Missing data
No imputation will be made for missing data, but quantities and location will inform future studies.
Data monitoring
The chief investigator will ensure access to source data and documents for trial related monitoring by a representative of the study sponsor. There are three planned monitoring visits agreed with the trial sponsor. As this is a small, investigator-initiated feasibility study, a data monitoring and ethics committee will not be convened. There is also no planned interim analysis, however interim data maybe provided to the trial steering committee at their request.
Ethics & dissemination
Consent
Septic shock is a life-threatening emergency requiring urgent therapies such as fluid resuscitation, to maximise chances of patient survival. Patients with septic shock require fluid resuscitation immediately, but their physiological condition will likely render them unable to provide informed consent prior to enrolment. This, alongside the potential distress of the emergency situation, makes any attempt to obtain either prior informed consent from the patient, or advice from their Personal Legal representative (i.e. relative or close friend), inappropriate. Considering these reasons, and with the support of our patient and public engagement group, once an eligible patient is identified for the trial, they will be enrolled and randomised as soon as possible (deferred consent).
Following enrolment through deferred consents a member of the research team will contact the patient’s personal legal representative to discuss the trial and seek advice on the patient’s likely views on participating in this trial. If the patient’s personal legal representative is unavailable or unwilling/unable to engage in these discussions, then written agreement from the patient’s professional legal representative will be gained instead. Written agreement from a legal representative will be obtained within 7 days of randomisation.
Once a patient has regained capacity, they will be approached by a member of the research team for informed deferred consent to continue participation. Patients who die prior to regaining capacity will not be able to consent to continue participation, however their data will be included in the trial analysis. If a patient is discharged from hospital prior to providing consent, they will be contacted by telephone and written letter requesting they complete the consent process. If there is no response, their data collected to date will be used during final analysis. Both patients and their legal representatives have the right to withdraw their consent for continued participation at any time.
Dissemination
This trial will be reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) guidelines. 11 Our findings will be presented at national and international meetings and summarised in a written manuscript for publication in a peer reviewed medical journal. Patients involved in the trial and members of our patient engagement group will be provided with a lay summary of the principal study findings either by post or e-mail depending on their preference.
Discussion
The existing evidence for the use of albumin in critical care identifies those with septic shock as the most likely to benefit and worthy of further study. Hyperoncotic albumin is associated with lower cumulative fluid balance, faster resolution of shock and has been used independently as both a resuscitation fluid and regular supplement.
This trial will objectively test the feasibility of utilising hyperoncotic albumin combined as a resuscitation fluid and daily supplement in patients with early septic shock. We have pre-specified thresholds for feasibility outcomes including recruitment rate, protocol adherence, data capture and safety. The robust data acquired from this study will subsequently support or refute the viability of a future multicentre, cost effectiveness trial. All regulatory approvals have been obtained and the trial is open to recruitment since November 2023. Recruitment is expected to be complete within 24 months.
Footnotes
Author’s note

| Version 2.2, dated 14/03/2024 | |
|---|---|
| Trial acronym | SWIPE 2 |
| IRAS Project ID | 1005399 |
| ClinicalTrials.gov identifier | NCT05208242 |
| EudraCT No. | 2022-000997-24 |
Author’s contributions
JBS and RB conceived the study. JBS developed study design and drafting of this protocol with advice from RB, TF, DM, & PD. CF and JBS will complete all statistical analyses. GK will assist with analysis of plasma samples from the study biobank. AT will assist with analysis of health economic data.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JBS has previously received an unrestricted research grant and free albumin from CSL Behring to support another trial. 7 RB has received research funding from CSL Behring in support of the HAS Flair II trial. 12
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is funded through a grant from Manchester Academic Health Sciences Centre through Health Innovation Manchester. CSL Behring provided 20% albumin free of charge for all participants in the intervention arm. Neither supporting institutions had any influence on the design of this protocol, nor conduct of the trial.