
Abstract
Vasodilatory shock is common in critically ill patients and vasopressors are a mainstay of therapy. A meta-analysis suggested that use of a higher, as opposed to a lower, mean arterial pressure target to guide titration of vasopressor therapy, could be associated with a higher risk of death in older critically ill patients. The 65 trial is a pragmatic, multi-centre, parallel-group, open-label, randomised clinical trial of permissive hypotension (a mean arterial pressure target of 60–65 mmHg during vasopressor therapy) versus usual care in critically ill patients aged 65 years or over with vasodilatory hypotension. The trial is conducted in 2600 patients from 65 United Kingdom adult, general critical care units. The primary outcome is all-cause mortality at 90 days. An economic evaluation is embedded. The 65 trial received favourable ethical opinion from the South Central – Oxford C Research Ethics Committee and approval from the Health Research Authority. The results will be presented at national and international conferences and published in peer-reviewed medical journals.
Trial registration: ISRCTN10580502
Background
Vasodilatory shock, a clinical syndrome of which hypotension is a cardinal feature, is common in critically ill patients. While increasing blood pressure is a complex process, vasopressors – intravenous medications that increase blood pressure by causing vasoconstriction – are a mainstay of therapy.
United Kingdom (UK) national clinical audit data indicate that close to half (44%) of all patient admissions to adult, general critical care units have hypotension and receive vasopressor therapy and it is estimated that 40–50% of these cases will represent vasodilatory hypotension. Clinicians give vasopressors to avoid hypotension; however, excessive vasoconstriction and other effects associated with vasopressors – such as increased cardiac workload – could actually cause harm to critically ill patients. Clinicians in critical care units are therefore faced with the challenge of balancing the risks of hypotension with the risks of vasopressor-associated side effects.
To guide treatment with vasopressors, doctors typically prescribe a mean arterial pressure (MAP) target and bedside nurses continuously adjust the rate of vasopressor infusions to achieve the target MAP. Previous guidelines recommended maintaining MAP above 65 mmHg, but these were based on low quality evidence and did not provide guidance for an upper MAP limit. 1 Since the optimal blood pressure target is not well established, clinicians tend to err on the side of maintaining MAP well above recommended thresholds. 2 This practice, which exposes patients to vasopressor-induced side effects, may be especially problematic in older, more vulnerable critically ill patients. A pooled analysis of two randomised clinical trials (RCTs)3,4 suggested that, with increasing age, higher MAP targets may be associated with a greater risk of death.5,6
An alternative strategy is to target blood pressure values below traditional levels to reduce the exposure to vasopressors and minimise their side effects. This approach, termed permissive hypotension, echoes other strategies used in oxygen therapy, 7 enteral feeding, 8 mechanical ventilation, 9 blood transfusions, 10 intravenous fluids for patients after trauma 11 and severe febrile illness in children to minimise unnecessary critical care interventions including potential associated side effects. 12
Aim
The primary aim of the 65 trial is to evaluate the clinical and cost-effectiveness of permissive hypotension (MAP target range 60–65 mmHg during vasopressor therapy) in critically ill patients aged 65 years or over with vasodilatory hypotension. Our hypothesis is that the benefits associated with a reduced exposure to vasopressors outweigh the risks associated with lower MAP values.
Methods/design
Efficient design and setting
The 65 trial is a pragmatic, multi-centre, parallel-group, open-label, RCT of permissive hypotension versus usual care in critically ill patients aged 65 years or over with vasodilatory hypotension.
The 65 trial was designed to minimise the impact that research can create for critical care unit teams. The trial is nested in an existing network of research-active critical care units who participate in the case mix programme (CMP). The CMP, coordinated by the Intensive Care National Audit & Research Centre (ICNARC), is the national clinical audit for adult critical care in England, Wales and Northern Ireland and is a source of high quality, robust and representative data. The trial is enrolling eligible patients across 65 adult, general critical care units (‘sites’) who participate in the CMP.
Patient flow
Screening
Potentially eligible patients accepted for admission to the participating adult, general, critical care unit are screened against the inclusion/exclusion criteria by the local clinical team, supported by the site research team. Screening and Enrolment Logs record enrolled patients, reasons for exclusion and the reason eligible patients are not enrolled.
To be eligible for the 65 trial, patients must meet all eligibility criteria:
Inclusion criteria
Age 65 years or older. Vasodilatory hypotension as assessed by treating clinician. Started infusion (for at least 1 h) of vasopressors within prior 6 h (if noradrenaline, then a minimum dose of 0.1 µg kg−1 min−1). Adequate fluid resuscitation is completed or ongoing. Vasopressors expected to continue for 6 h or more as assessed by treating clinician.
Exclusion criteria
Vasopressors being used solely as therapy for bleeding, acute ventricular failure (left or right) or post-cardiopulmonary bypass vasoplegia. Ongoing treatment for brain injury or spinal cord injury. Death perceived as imminent. Previous enrolment to the 65 trial.
Previously, patients were eligible in whom a decision to start vasopressors (at any dose) had been made. The inclusion criteria were updated in December 2017 following routine central monitoring of available trial data for 159 control group patients which identified a group of patients who received a relatively short duration (and often low doses) of vasopressors only. The inclusion criteria were therefore refined to specify that, at the time of randomisation, patients must have been on a vasopressor infusion for at least 1 h and if receiving noradrenaline, then they must be on a dose of at least 0.1 µg kg−1 min−1.
Randomisation
Randomisation is concealed and performed as soon as possible after confirming eligibility. Patients are randomised following a 1:1 sequence to either the intervention group (permissive hypotension) or control group (usual care) using a continuously available dedicated telephone or web-based randomisation service. Allocation is stratified by recruiting site using permuted blocks with variable block lengths. As this is a large trial, the risk of chance imbalance in prognostic factors is low and the need to randomise patients during a very short time-frame mandates that the randomisation process is as simple as possible. For these reasons, we elected not to stratify the randomisation process on additional potential confounders.
Following randomisation, each participant is assigned a unique 65 Trial Number and a Case Report Form (CRF) to be completed by the local research team. The full flow of patients through the trial is shown in Figure 1.
The 65 trial patient flow.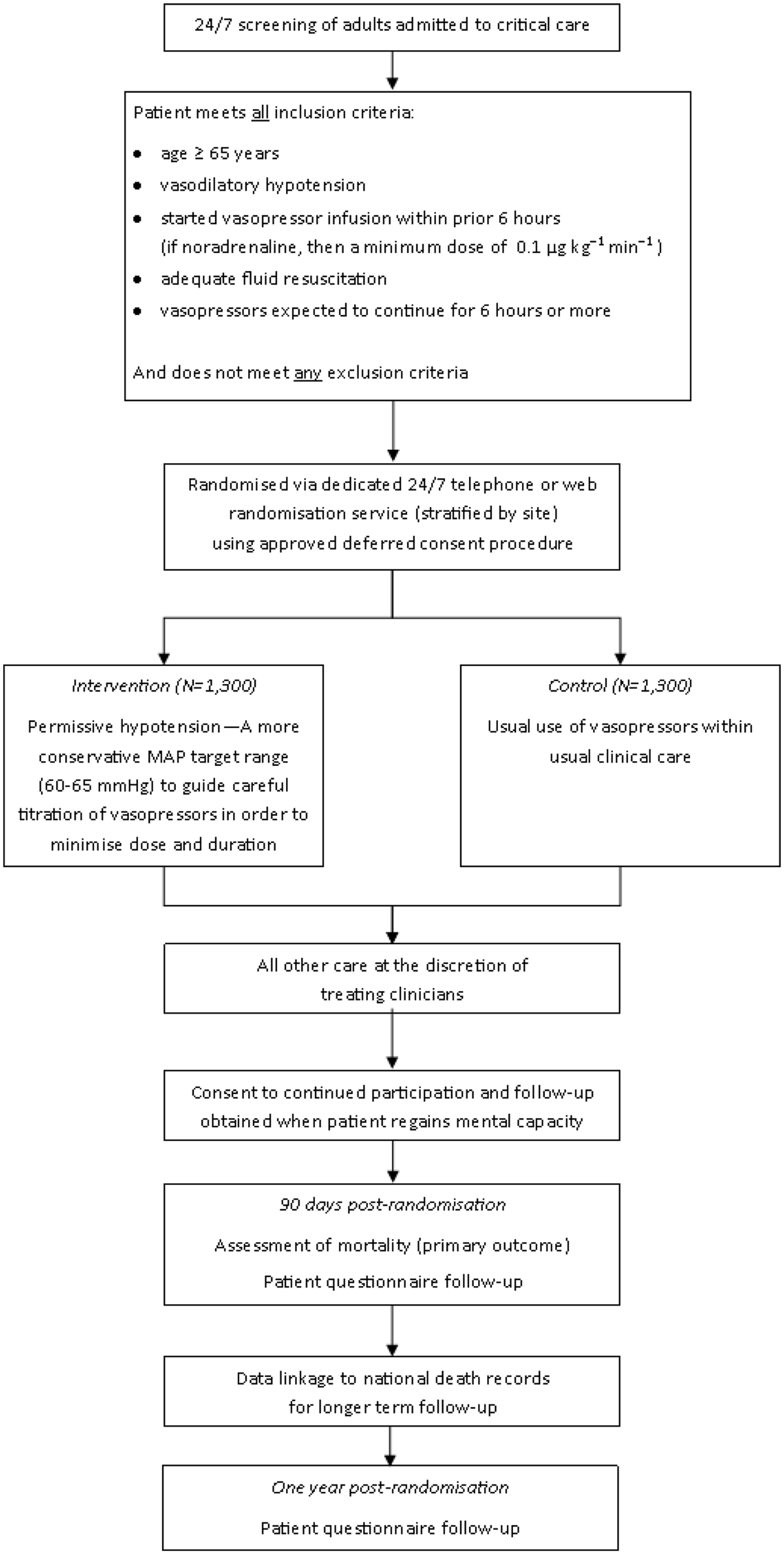
Trial interventions
Intervention group
Patients randomised to the intervention group will be treated using the permissive hypotension strategy – a MAP target range of 60–65 mmHg during vasopressor therapy. The permissive hypotension protocol begins immediately following randomisation. As soon as treatment allocation is disclosed, the staff member who randomised the patient should notify the medical team to prescribe a MAP target range of 60–65 mmHg. The clinical team adjusts the vasopressor infusions to maintain the patient's MAP within the prescribed range for as long as necessary until the patient is able to maintain the target MAP without vasopressors. The choice of vasopressor agent is left to the discretion of the medical team and should be administered as per local practices and guidelines. The following list defines vasopressors in the 65 trial:
Noradrenaline (Norepinephrine). Vasopressin. Terlipressin. Phenylephrine. Adrenaline (Epinephrine). Dopamine. Metaraminol.
The decision to discontinue vasopressors will depend on the patients' ability to maintain the MAP target stipulated by the protocol without vasopressors. The trial treatment will apply at any point the patient requires vasopressors during their admission in the critical care unit.
Control group
Patients in the control group will receive usual care (as per local practices).
Co-interventions
The use of inotropes, fluids and corticosteroids will be recorded but left to the discretion of the treating team.
Recruitment and consent
Patients who require vasopressors in critical care units often need this treatment started as a life-saving measure during an emergency situation. They usually also lack mental capacity to provide informed consent due to their medical condition and the effects of treatments administered as part of standard care (e.g. mechanical ventilation, sedative and analgesic drugs, etc.). In such an emergency situation, any delay in commencing the delivery of the trial intervention would be detrimental to the scientific validity of the trial. This, alongside the potential distress of the emergency situation, renders attempts to obtain informed consent from the patient, or an opinion from their Personal Consultee (i.e. relative or close friend), prior to starting the trial treatment inappropriate.
Considering these reasons, eligible patients are enrolled and randomised to receive the assigned treatment immediately. This approach is known as ‘deferred consent’ or ‘research without prior consent’ and is covered by an emergency waiver of consent, approved by the research ethics committee, under the Mental Capacity Act.
If patients do not regain capacity in a day or two after randomisation, a delegated member of the site research team approaches the patient's Personal Consultee as soon as appropriate and practical to discuss the trial and to seek their opinion as to the patients' likely wishes and feelings regarding participating in research. Ideally, this discussion takes place within 24–48 h of randomisation, once the patient's medical situation is no longer an emergency. If a Personal Consultee advises that, in their opinion, the patient would not choose to participate in research, the trial treatment (if permissive hypotension) is immediately stopped and the Personal Consultee asked whether, in their opinion, the patient would consent to ongoing data collection and/or scheduled 65 trial questionnaire follow-up at three months and one year. Upon recovery, the patient is then approached directly for informed retrospective consent. Where the patient's decision is different to that of their personal consultees, the patient's decision is final and will supersede the Personal Consultee opinion.
Patients who have full capacity and are able to give informed consent prior to randomisation are asked to give verbal consent, followed by full written informed consent later.
If a patient dies, a Nominated Consultee is appointed. The Nominated Consultee can include an Independent Mental Capacity Advocate appointed by the NHS Hospital Trust or an independent doctor (i.e. not associated with the conduct of the trial). The opinion of the Nominated Consultee is sought in the same manner as for the Personal Consultee. A Nominated Consultee is also approached in the rare situations where no Personal Consultee is available.
If the patient is discharged from hospital with mental capacity prior to their consent decision being confirmed, then the patient is contacted by telephone and post to provide informed consent. The same process will be used if the patient is discharged without capacity, but with the patients Personal Consultee. If the participant is transferred to another hospital participating in the 65 trial before the consent procedures are complete, then the local research team contacts the research team at the receiving hospital to handover the consenting procedures.
Refusal or withdrawals of consent/opinion
If patient informed consent (or consultee opinion) is refused or withdrawn, this decision is respected and abided by, and no further contact made. All data up to the point of this decision are retained in the trial records, unless the patient or consultee requests otherwise.
Safety monitoring
All patients eligible for the 65 trial are critically ill, and due to the complexity of their condition are at increased risk of experiencing adverse events and serious adverse events (SAEs).
13
In the 65 trial, the labelling of a SAE is limited to serious events, which might reasonably occur as a consequence of either lower MAP values and/or higher doses of vasopressors required to maintain higher MAP values. In addition to reporting any unexpected and possibly related SAEs, research teams are asked to screen for the following events (up to critical care unit discharge) which are included in the CRF:
Supraventricular cardiac arrhythmia. Ventricular cardiac arrhythmia. Myocardial infarction. Extremity necrosis. Mesenteric ischaemia. Severe acute renal failure.
Each event is assessed for its severity, using the below scale:
None: indicates no event or complication. Mild: complications result in only temporary harm and do not require clinical treatment. Moderate: complications require clinical treatment but do not result in significant prolongation of hospital stay. Does not usually result in permanent harm and where this does occur the harm does not cause functional limitations to the patient. Severe: complications require clinical treatment and result in significant prolongation of hospital stay and/or permanent functional limitation. Life threatening: complications may lead to death. Fatal: indicates that the patient died as a direct result of the complication/adverse events.
A reportable event with the severity of ‘severe’, ‘life-threatening’ of ‘fatal’ is considered an SAE in the 65 trial and is reported on the SAE Reporting Form. On receipt of an SAE report, a clinical member of the 65 Trial Management Group evaluates the event for relatedness and expectedness to determine whether or not the case qualifies for expedited reporting to the Research Ethics Committee. If the event is judged unexpected and potentially related to the trial interventions, the ICNARC CTU submits a report to the REC within 15 calendar days.
Follow-up
For the primary endpoints, patients are followed up until day 90. Thereafter, each participant is followed up with a questionnaire up to a maximum of one year. At each time-point (day 90 and one year), survivors receive a postal questionnaire containing the EuroQol EQ-5D-5L questionnaire, 14 Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE, short version) 15 and a health services use questionnaire. The one-year questionnaire follow-up is curtailed and will end once the last patient has completed their 90-day questionnaire. Non-responders are telephoned and offered various completion options, including completing the questionnaire over the telephone.
Outcomes
Primary outcome – Clinical effectiveness:
All-cause mortality at 90 days.
Primary outcome – Cost-effectiveness:
Incremental net monetary benefit (INB), evaluated at the National Institute for Health and Care Excellence (NICE) recommended threshold of £20,000 per quality-adjusted life year (QALY), at 90 days.
Secondary outcomes:
Mortality at discharge from the critical care unit and acute hospital. Duration of survival to longest available follow-up. Duration of advanced respiratory and renal support (defined according to the UK Department of Health Critical Care Minimum Dataset) during the critical care unit stay. Days alive and free of advanced respiratory support and renal support within first 28 days. Duration of critical care unit and acute hospital stay. Cognitive decline assessed using the Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE, short version) at 90 days and one year. Health-related quality of life, assessed using the EuroQol EQ-5D-5L questionnaire, at 90 days and one year. Resource use and costs at 90 days and one year. Estimated lifetime incremental cost-effectiveness.
Data collection
Patient data collection schedule.
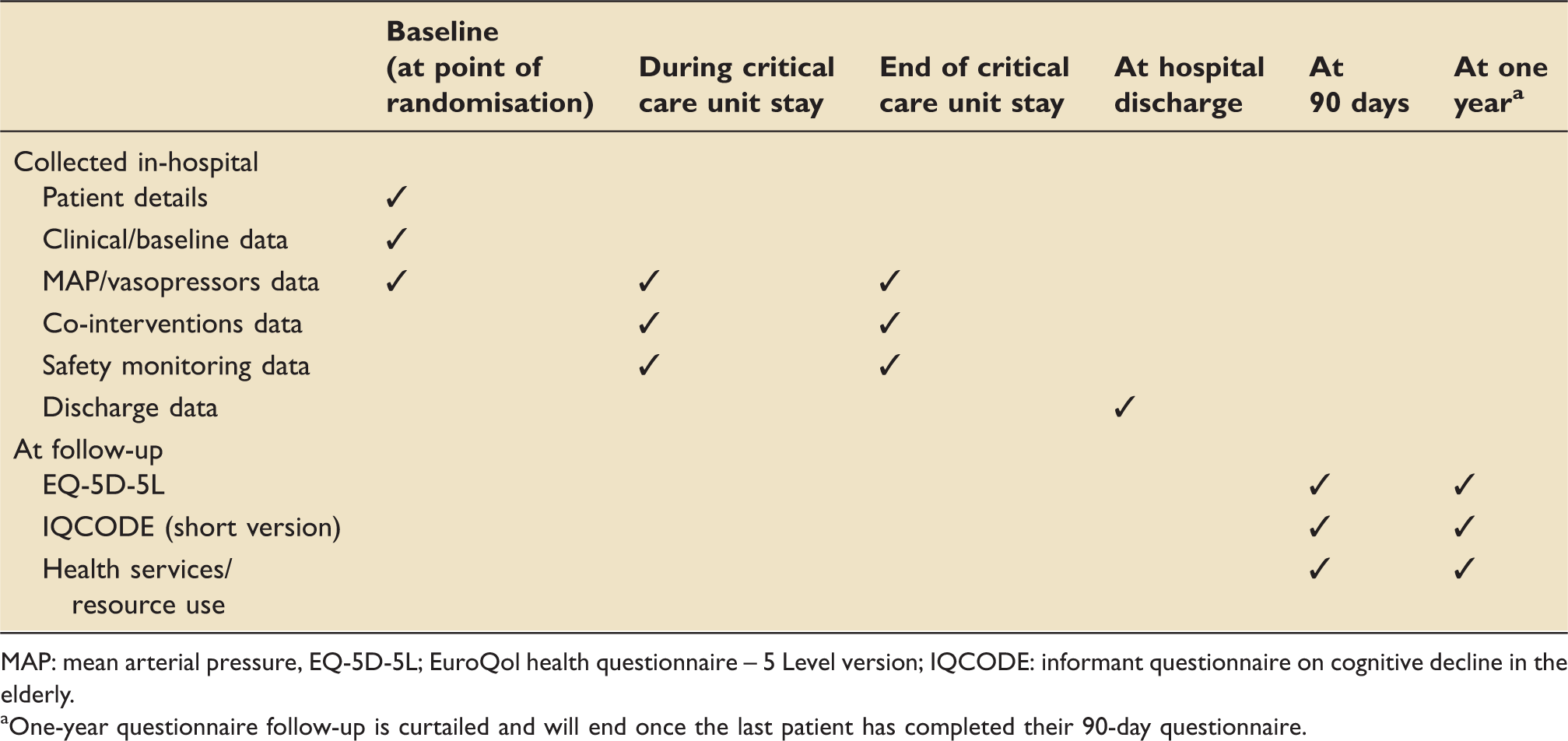
MAP: mean arterial pressure, EQ-5D-5L; EuroQol health questionnaire – 5 Level version; IQCODE: informant questionnaire on cognitive decline in the elderly.
One-year questionnaire follow-up is curtailed and will end once the last patient has completed their 90-day questionnaire.
Nesting the 65 trial within the CMP provides further rich data on demographics, surgical status, acute severity of illness, duration of organ support and duration of critical care unit stay. Linking the trial data to civil registration data will provide mortality data up to the point that the database is locked (including the primary outcome at 90 days). This latter data linkage will be in accordance with a Data Sharing Agreement between ICNARC and NHS Digital.
Identifiable patient data, including full name, contact details, date of birth and NHS number are required by the ICNARC CTU to successfully follow-up participants and to link to the above detailed data sources. The ICNARC CTU acts to preserve participant confidentiality and all data are stored securely, managed in accordance with ICNARC CTU Standard Operating Procedures and the Data Protection Act (2018).
Protecting against sources of bias
Selection bias is minimised by the concealment of allocation through a centrally administered randomisation system. While a method for double-blinded blood pressure target investigation has been reported, 16 we felt this would be impractical in a large pragmatic trial. To reduce the risk of performance bias, information on the use of relevant cointerventions is collected daily on the 65 trial CRF. To reduce the risk of ascertainment bias for secondary endpoints (e.g. safety outcomes such as severe acute renal failure requiring renal replacement therapy), key endpoints will also be collected from the CMP using data linkage. To reduce the risk of bias associated with loss to follow-up, the primary outcome will be ascertained using multiple sources including the participants' medical records and through data linkage to the CMP and civil registration data. Primary analyses will adhere to the intention-to-treat (ITT) principle.
Power calculation
Assuming 90-day mortality of 35% within usual care (control group – based on CMP data for patients aged 65 years or older admitted to critical care and receiving advanced cardiovascular support defined according to the Critical Care Minimum Dataset) and a 2.5% withdrawal/loss to follow-up rate, a sample size of 2600 patients (1300 per group) will provide 90% power to detect as statistically significant (P < 0.05) a 6% absolute risk reduction (ARR) – corresponding to a 17% relative risk reduction (RRR) – to 29% in the intervention group.
If the pre-trial assumption regarding the control group event rate is incorrect, this sample size will retain at least 85% power to detect the specified ARR (and smaller RRR) even if the mortality is as high as 50% and at least 80% power to detect the specified RRR (and smaller ARR) if the mortality is as low as 29%.
It is anticipated that recruitment will be completed by 65 sites recruiting for approximately 21 months (accounting for staggered activation of sites).
A previous power calculation, which specified an absolute risk reduction of 8% between groups and which required 1440 patients (720 in each arm), was updated following the internal pilot phase. This change was recommended by the Trial Steering Committee (TSC) after the internal pilot identified that the duration of vasopressor therapy in the control group was lower than initially expected, suggesting that the difference in treatment (and hence outcome) between arms may be smaller than initially anticipated.
Statistical analysis
Internal pilot analysis
An internal pilot was conducted on patients recruited during the first six months (internal pilot phase). The anticipated sample size at this point provided 99% power to detect as statistically significant (P < 0.05) the pre-specified clinically important separation between groups both of 10 mg (norepinephrine equivalent) in mean total vasopressor dose, assuming a standard deviation of 15 mg in each group, and a separation of 5 mmHg in peak MAP while receiving vasopressors, assuming a standard deviation of 7.5 mmHg in each group. The secondary pilot objectives were to open a minimum of 50 sites and for the recruitment rate in open sites to be at least 80% of anticipated. All pilot objectives were met.
Interim analyses
The number of interim analyses was limited to detect early evidence of harm and irrefutable mortality differences. A single interim analysis was carried out after the recruitment and follow-up of 500 patients using a Peto-Haybittle stopping rule (P < 0.001) to recommend early termination due to either effectiveness or harm. Following the planned interim analysis, the Data Monitoring and Ethics Committee (DMEC) recommended the continuation of the trial with no changes.
Clinical effectiveness analysis
All analyses will be lodged in a statistical analysis plan,17 a priori, before the investigators are unblinded to any outcomes. All analyses will be performed according to the ITT principle and the trial will be reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) statement. 18
Analysis of dichotomous outcomes will be performed both unadjusted (using Fisher's exact test) and adjusted for baseline covariates (using multilevel logistic regression with unit-level random effects). Analyses of time-to-event data (time to death) will be performed by Kaplan–Meier methods and Cox proportional hazards modelling. Analyses of days alive and free of advanced respiratory and/or renal support will be performed by bootstrapped t-tests to account for non-normality. Analyses of duration of critical care unit and acute hospital stay will be performed by Wilcoxon rank-sum tests, stratified by survival status. Analyses of cognitive function and health-related quality of life will be performed by t-tests and adjusted linear regression. Subgroup analyses will be performed to test for interactions between the effect of allocated treatment group and the pre-specified baseline covariates.
Health economic evaluation
A full cost-effectiveness analysis will be undertaken to assess the relative cost-effectiveness of the intervention versus usual care. Resource use and outcome data collected as part of the trial will be used to report the relative cost-effectiveness at 90 days, according to the incremental net benefit, and to also project the lifetime cost-effectiveness.
The cost analysis will use detailed, micro-costing methods to record the costs of providing vasopressors within the critical care unit. This approach will enable the cost analysis to recognise any cost variation across different patient subgroups. Each patient's critical care unit admission will be assigned to the appropriate Healthcare Resource Group (HRG) using mandated data for the CCMDS. The cost per hospital bed-day for each HRG category for critical care, and for general medical bed-days will be available from the NHS Payment by Results database.
The cost analysis will take a health and personal health services perspective. The cost-effectiveness analysis will report the mean (95% confidence interval) incremental costs and QALYs of the intervention versus usual care at 90 days, incremental net benefit (INB) at a willingness to pay of £20,000 per QALY, and the probability that the intervention is cost-effective compared with usual care at different levels of willingness to pay for a QALY gained. The cost-effectiveness analysis will use regression methods to report relative cost-effectiveness according to pre-defined subgroups and will be combined with multiple imputations to address issues posed by missing EQ-5D-5L or cost data. Survival analysis will be used to extrapolate any within-trial differences in costs and QALYs in projecting lifetime cost-effectiveness.
Sensitivity analyses will test whether the results are robust to methodological assumptions.
Trial closure
The end of the trial will be when all participants have completed their 90-day follow-up, at which point the ‘Declaration of end of trial’ form will be submitted to the REC by the ICNARC CTU.
Ethical compliance
The 65 trial received favourable ethical opinion from the South Central – Oxford C Research Ethics Committee (reference: 17/SC/0142) and approval from the Health Research Authority (Integrated Research Application System (IRAS) reference: 215503) prior to the commencement of patient recruitment. Confirmation of capacity and capability (i.e. local approvals) was obtained from each participating site prior to patient recruitment.
The 65 trial is conducted in accordance with the approved trial Protocol, International Conference on Harmonisation – Good Clinical Practice (ICH-GCP) guidelines, the UK Policy Framework for Health and Social Care Research, the Data Protection Act (2018), the Mental Capacity Act in England and Wales (2005), as well as the ICNARC CTU research policies and procedures.
Trial management and oversight
The Trial Management Group (TMG) is responsible for management of the 65 trial and is led by PM (Chief Investigator) and FL (Lead Clinical Investigator) who take overall responsibility for delivery of the trial and oversee progress against timelines/milestones. The TMG also comprises methodological, clinical and PPI co-investigators as well as members of the ICNARC CTU trial team.
Both a Trial Steering Committee (TSC) and a Data Monitoring and Ethics Committee (DMEC) have been convened as trial oversight committees. The TSC is independently chaired by Professor Tim Walsh (University of Edinburgh), is made up of experienced clinicians, methodologists and PPI representatives, and takes responsibility for overall supervision on behalf of the Sponsor and Funder. The independent DMEC, chaired by Professor John Norrie (University of Edinburgh), monitors recruitment, protocol adherence and patient safety and includes experienced methodologists and clinicians.
ICNARC is Sponsor (contact details available at http://www.icnarc.org) for the 65 trial and holds professional indemnity insurance to meet the potential legal liability of the Sponsor and employees for harm to participants arising from the design and management of the research. Indemnity to meet the potential legal liability of investigators/collaborators for harm to participants arising from the conduct of the research is provided by the NHS indemnity scheme or through professional indemnity.
Dissemination
The results of the 65 trial will be widely and actively disseminated. Presentations will be scheduled for national and international critical care conferences and articles will be prepared for publication in peer-reviewed scientific journals, as well as in relevant professional journals.
Discussion
The 65 trial was informed by emerging evidence suggesting that minimising vasopressor exposure may save lives in older, critically ill patients. This multi-centre RCT is the first adequately powered trial to evaluate the clinical effectiveness and economic impact of a protocol aiming to reduce exposure to commonly used vasopressor medications. The importance of this research, for patients and for health systems, hinges on the enormous burden of vasodilatory hypotension the world over. Paradoxically, a very small number of patients have been enrolled in clinical trials comparing different intensity of vasopressor therapy in this context and older patients are even less likely to be enrolled in critical care trials. 19 In the critical care unit, historically, we cared for young and previously healthy patients who may have tolerated adverse effects of vasopressors. Today, and in the future, an increasing number of frailer, elderly patients, who are more sensitive to iatrogenic complications, will receive critical care. 20
Trial status
At the time of manuscript submission, patient recruitment for the trial is ongoing and planned to be complete in the first quarter of 2019. The first participant was recruited in July 2017. The trial results will be disseminated in early 2020 through publications in peer-reviewed scientific journals and presentations at national and international conferences.
Footnotes
Acknowledgements
The authors thank the following people for the contributions to the set-up and delivery of the 65 trial: Robert Darnell, Nick Hudson, Joseph Collins, Sian Martin, Abby Koelewyn, Laura Drikite, Akshay Patel, Karen Thomas and Michelle Saull. The authors also thank the research and clinical staff at all of the participating sites: Addenbrookes Hospital, Aintree University Hospital, Altnagelvin Hospital, Antrim Area Hospital, Arrowe Park Hospital, Basingstoke and North Hampshire Hospital, Blackpool Victoria Hospital, Bristol Royal Infirmary, Broomfield Hospital, Charing Cross Hospital, Countess of Chester Hospital, Darent Valley Hospital, Darlington Memorial Hospital, Derriford Hospital, Dorset County Hospital, Glangwili General Hospital, Gloucestershire Royal Hospital, Hammersmith Hospital, Ipswich Hospital, James Cook University Hospital, King's College Hospital, Leicester Royal Infirmary, Lister Hospital, Manchester Royal Infirmary, Medway Maritime Hospital, Morriston Hospital, Musgrove Park Hospital, Norfolk and Norwich Hospital, North Devon District Hospital, Northampton General Hospital, Northern General Hospital, Peterborough City Hospital, Pinderfields Hospital, Poole Hospital, Queen Alexandra Hospital, Queen Elizabeth Hospital (Gateshead), Queen Elizabeth Hospital (Woolwich), Queens Medical Centre, Royal Berkshire Hospital, Royal Blackburn Hospital, Royal Cornwall Hospital, Royal Devon and Exeter Hospital, Royal Glamorgan Hospital, Royal Gwent Hospital, Royal Liverpool University Hospital, Royal Oldham Hospital, Royal Preston Hospital, Royal Stoke University Hospital, Royal Victoria Infirmary, Russells Hall Hospital, Salford Royal Hospital, Southmead Hospital, St Mary's Hospital (London), St Thomas' Hospital, The Princess Royal University Hospital (Farnborough), Torbay Hospital, Tunbridge Wells Hospital, University Hospital Coventry, University Hospital Lewisham, University Hospital of North Tees, Warwick Hospital, William Harvey Hospital, Worthing Hospital, Yeovil District Hospital and York Hospital.
Authors' contributions
PRM is Chief Investigator. FL is Lead Clinical Investigator. ARB is Trial Manager. KMR is the director of the Clinical Trials Unit and has oversight of the trial. JC, ACG, RG, DHa, DHe, ZS, CW and DY are co-investigators and co-applicants on the grant. FL, ARB and PM drafted the manuscript and all authors read and approved the final version.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: ACG reports that outside of this work he has received speaker fees from Orion Corporation Orion Pharma and Amomed Pharma. He has consulted for Ferring Pharmaceuticals, Tenax Therapeutics, Baxter Healthcare, Bristol-Myers Squibb and GSK, and received grant support from Orion Corporation Orion Pharma, Tenax Therapeutics and HCA International with funds paid to his institution. All other authors declare no conflict of interests.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was funded by the National Institute for Health Research (NIHR) Health Technology Assessment (HTA) Programme (project number: 15/80/39). ACG is funded by an NIHR Research Professor award (RP-2015-06-018) and by the NIHR Imperial Biomedical Research Centre. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the HTA Programme, NIHR, NHS or the Department of Health and Social Care.
Research ethics and patient consent
This paper represents Protocol version 3.1 (dated 20 February 2019) which received favourable ethical opinion from the South Central – Oxford C Research Ethics Committee (REC) (reference: 17/SC/0142) and approval from the Health Research Authority. The full Protocol (including amendments) is available on the NIHR website. The 65 trial was granted an emergency waiver of consent, with patients and/or their Consultee (as appropriate) approached for consent when appropriate following randomisation.