
Abstract
One of the few interventions to demonstrate improved outcomes for acute hypoxaemic respiratory failure is reducing tidal volumes when using mechanical ventilation, often termed lung protective ventilation. Veno-venous extracorporeal carbon dioxide removal (vv-ECCO2R) can facilitate reducing tidal volumes. pRotective vEntilation with veno-venouS lung assisT (REST) is a randomised, allocation concealed, controlled, open, multicentre pragmatic trial to determine the clinical and cost-effectiveness of lower tidal volume mechanical ventilation facilitated by vv-ECCO2R in patients with acute hypoxaemic respiratory failure. Patients requiring intubation and mechanical ventilation for acute hypoxaemic respiratory failure will be randomly allocated to receive either vv-ECCO2R and lower tidal volume mechanical ventilation or standard care with stratification by recruitment centre. There is a need for a large randomised controlled trial to establish whether vv-ECCO2R in acute hypoxaemic respiratory failure can allow the use of a more protective lung ventilation strategy and is associated with improved patient outcomes.
Keywords
Introduction
Background and rationale
Acute hypoxaemic respiratory failure requiring mechanical ventilation is a major cause of morbidity and mortality. A significant proportion of affected patients will meet the diagnostic criteria for the Acute Respiratory Distress Syndrome (ARDS). 1 Acute hypoxaemic respiratory failure and ARDS occur in response to a variety of insults, have a mortality of approximately 40%, cause a long-term reduction in quality of life for survivors, and have significant resource implications.2–4
Mechanical ventilation may be required as an immediate life saving intervention, but is also known to contribute to the morbidity and mortality. 5 Exposing the injured lung to the high “driving” pressures and tidal volumes from mechanical ventilation may cause regional over-distension resulting in further ventilator-associated lung injury (VALI). The few interventions that have been shown to reduce the high mortality in patients with acute hypoxaemic respiratory failure and ARDS have reduced injurious ventilation.6,7 A landmark trial found that mechanically ventilating patients with acute hypoxaemic respiratory failure secondary to ARDS with a lung protective strategy aiming for a tidal volume of 6 ml/kg predicted body weight (PBW) and a plateau pressures less than 30 cm/H20, decreased mortality from 40% to 31%.8,9
Recent studies have shown that lung hyperinflation and injury still occur in many patients with ARDS, despite ventilation with what would be considered as “protective”. 10 It is biologically plausible that further tidal volume reduction may be beneficial, and this is supported by pilot clinical data. 11 However, further tidal volume reduction can be associated with secondary systemic effects associated with raised blood carbon dioxide levels, such as elevated intracranial pressure, pulmonary hypertension, altered myocardial contractility and decreased renal blood flow.
Extracorporeal carbon dioxide removal (ECCO2R) in association with very low tidal volume mechanical ventilation offers a potentially attractive solution to reduce VALI further, whilst mitigating these potential adverse effects. 12 Using ECCO2R, carbon dioxide can be removed from the blood while the lungs are ventilated in a more protective manner. In recent years, more efficient veno-venous (vv-ECCO2R) devices have become available, replacing arterio-venous devices and the need for arterial puncture. Such systems can achieve carbon dioxide removal with relatively low extracorporeal blood flows (0.4−1 L/min), and require only a single dual lumen venous catheter comparable to renal dialysis, which is routinely used safely as standard care in ICUs in the UK.
A recently completed systematic review to assess feasibility, complication rates, and efficacy of ECCO2R devices in acute respiratory failure and ARDS included 14 studies with 495 patients (two randomised controlled trials and 12 observational studies). 13 Overall, there was a paucity of high quality data. Carbon dioxide removal was shown to be feasible, facilitating the use of lower tidal volume ventilation. There was no survival benefit demonstrated with ECCO2R, although a post hoc analysis of data from the most recent randomised controlled trial showed an improvement in ventilator-free days in patients with more severe hypoxia (as defined as a ratio of the partial pressure of oxygen in arterial blood over the fractional inspired oxygen (PaO2/FiO2) ratio of less than 20 kPa). 14 While these data suggest improved outcomes (as indicated by more ventilator free days) with the application of ECCO2R in patients with more severe respiratory failure, definitive data are as yet lacking. The systematic review demonstrated clinical equipoise on the role of vv-ECCO2R in acute respiratory failure.
As the technology to deliver vv-ECCO2R has become simpler, it is increasingly being adopted into clinical practice. There is thus a need for a large randomised controlled trial to determine whether vv-ECCO2R in acute hypoxaemic respiratory failure can allow the use of a more protective ventilatory strategy and is associated with improved patient outcomes.
Objectives
Primary objective
To determine whether vv-ECCO2R and lower tidal volume mechanical ventilation in patients with acute hypoxaemic respiratory failure decreases mortality 90 days after randomisation compared with standard care.
Secondary objectives
In mechanically ventilated patients with acute hypoxaemic respiratory failure to determine the effects of vv-ECCO2R on tidal volumes, duration of mechanical ventilation, requirement for extracorporeal membrane oxygenation (ECMO), long-term mortality, health-related quality of life, safety, cost-effectiveness, and long-term morbidity.
Methods
This manuscript was written in concordance with the SPIRIT guidelines. 15
Trial design
This is a multicentre, randomised, allocation concealed, controlled, open, pragmatic clinical and cost-effectiveness trial. The main trial will be preceded by a six-month internal pilot study to test recruitment and compliance with assigned treatment. The trial will be conducted and managed by the Northern Ireland Clinical Trials Unit (NICTU) and sponsored by the Belfast Health and Social Care Trust. The funding is provided by the National Institute for Health Research (NIHR) following a commissioned call from the Health Technology Assessment programme (HTA study reference 13/143/02). The trial will be conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice. The vv-ECCO2R devices and consumables will be provided free of charge by the manufacturer, Alung® Technologies (Pittsburg, PA, USA). Alung® Technologies had no role in the study design and protocol development and will have no role in the study conduct, data analysis, or data interpretation.
Participants, interventions and outcomes
Study setting
The main trial will take place in at least 40 ICUs. 16 The ICUs must provide evidence that they have a proven track record of participating in ICU research, have access to the relevant population and consultants in the ICU who have clinical equipoise for vv-ECCO2R in this setting and agree to maintain trial allocation in patients randomised by their colleagues. Many sites will have limited experience with the management of vv-ECCO2R, therefore access to an educational package addressing catheter insertion and maintenance of the device will be provided.
Eligibility criteria
Inclusion criteria
Patients will be included who:
are receiving invasive mechanical ventilation using positive end expiratory pressure (PEEP) greater than or equal to 5 cmH2O; have an acute and potentially reversible cause of hypoxaemic respiratory failure as determined by their treating physician; are within 48 h of the onset of hypoxaemia as defined by a PaO2/FiO2 ratio of less than or equal to 20 kPa.
Exclusion criteria
Patients will be excluded if they meet one or more of the following criteria:
age less than 16 years old; intubated and mechanically ventilated via an endotracheal or tracheostomy tube greater than or equal to 7 days (168 h) up to the time of randomisation; ability to maintain tidal volumes less than or equal to 3 ml/kg predicted body weight (PBW) while maintaining arterial blood pH above 7.2 as determined by the treating physician; receiving, or decision to commence ECMO in the next 24 h; mechanical ventilation using high frequency oscillatory ventilation or airway pressure release ventilation; untreated pulmonary embolism, pleural effusion or pneumothorax as the primary cause of acute respiratory failure; acute respiratory failure fully explained by left ventricular failure or fluid overload (determined by clinical assessment or echocardiography/cardiac output monitoring); left ventricular failure requiring mechanical circulatory support; contra-indication to limited systemic anticoagulation with heparin; unable to obtain vascular access to a central vein (internal jugular or femoral vein); inferior vena cava filter (if planning to use a femoral vein catheter); consent declined; treatment withdrawal expected within 24 h; patients not expected to survive six months on basis of premorbid health status; do not attempt resuscitation order in place; severe chronic respiratory disease requiring domiciliary ventilation (except for sleep disordered breathing); severe chronic liver disease (Child Pugh score greater than 11); platelet count less than 40,000 mm3; previously enrolled in the REST trial; prisoners.
The inclusion and exclusion criteria are designed to include those who reflect the general population of critically ill patients with acute hypoxaemic respiratory failure who may benefit from the therapeutic intervention and exclude patients who are unlikely to benefit owing to their underlying condition or increased risk of a complication from vv-ECCO2R.
Intervention
In the intervention arm, a single dual lumen catheter will be inserted into a central vein by an appropriately trained clinician. Vv-ECCO2R will then commence. Tidal volumes will be reduced incrementally aiming for a tidal volume less than or equal to 3 ml/kg PBW. The intervention will continue for at least 48 h after which patients will be weaned off vv-ECCO2R as per study manual. ECCO2R will be used for a maximum of seven days as part of the study protocol.
The device will be weaned when patients are less hypoxaemic and have demonstrated signs of clinical improvement. The duration of this may vary between patients.
Standard care
When randomised to standard care, it is recommended that patients are mechanically ventilated according to best practice using a tidal volume of 6 ml/kg PBW. 8
Patients in both the intervention and control arms can receive neuromuscular blocking drugs (NMBD) at any stage as clinically indicated. 17 Interventions including prone positioning or referral for consideration of ECMO can be applied in either arm of the trial as per standard care in the UK.18,19 The crossover and use of ECCO2R in the non-interventional arm will be considered a protocol violation.
Outcome measures
Primary outcome measure
All cause mortality 90 days after randomisation
Secondary outcome measures
Tidal volume (ml/kg PBW) at day 2 and day 3 after randomisation Ventilator free days at 28 days after randomisation Duration of ventilation in survivors after randomisation at 28 days; Need for ECMO up to day 7; Mortality rate at 28 days, six months and one year after randomisation Health-Related Quality of Life (HRQoL) at six months and one year after randomisation Adverse event rate; Health and Social Care Service costs at six months and one year; St. George’s Respiratory Questionnaire (SGRQ) at one year and need for home oxygen at six months and one year after randomisation Post Traumatic Stress Syndrome Questionnaire (PTSS-14) at one year after randomisation Incidence of post traumatic stress disorder and cognitive dysfunction
Exploratory outcome measure
Right heart function as determined by echocardiography during 6 ml/kg PBW and ≤3 ml/kg PBW tidal volume ventilation.
Sample size
The required sample size is 1120 patients. With 90% power at a p-value of 0.05 with an estimated 3% dropout due to loss to follow-up or withdrawal of consent), 560 per group will be required to detect a 23% relative reduction (9% absolute reduction) in 90-day mortality, assuming a control group mortality of 41%. The ARDSNet ARMA trial demonstrated a 9% absolute risk reduction in patients with hypoxaemic respiratory failure secondary to ARDS with lung protective ventilation. 8 This sample size would also detect a 20% relative reduction (8% absolute reduction) at 80% power. The control group mortality estimate is based on 2 sources. The first was the control group mortality of the most recent and largest trial in a similar cohort of patients with respiratory failure in intensive care 20 and the second is from a group of patients with similar degree of hypoxaemia from the UK Intensive Care National Audit and Research Centre database. These data are also supported by the global LUNGSAFE study. 21
Assignment of interventions
Randomisation
Patients will be randomised using an online or telephone centralised randomisation facility. Randomisation will be stratified by recruitment centre.
Participants will be allocated to vv-ECCO2R with lower tidal volume mechanical ventilation or standard care on a 1:1 ratio. If allocated to vv-ECCO2R, this should ideally be commenced within 8 h of randomisation.
Blinding
By the nature of the intervention, it will not be possible to blind whether a participant has been randomised to vv-ECCO2R or standard care.
Data collection, management and analysis
Data collection
Data will be entered via an electronic case report from (eCRF). Clinical data will be collected during the ICU stay up to 28 days after randomisation. This is outlined in the study schematic in Figure 1 and detailed in Table 1. Health-related quality of life and Health & Social Care Service use will be assessed at six months and one year after randomisation. The EQ-5D-5L is a generic preference-based measure of health that is recommended by NICE for economic evaluations. A resource use questionnaire will be administered at six months and one year after randomisation and will capture the patients’ use of health services, such as community, social, hospital and care services as well as oxygen usage. Patients will also be required to complete the St George Respiratory Questionnaire (SGRQ) one year after hospital discharge. The SGRQ has been used for a range of respiratory disease groups and is responsive to therapeutic trials as a gauge of respiratory disease burden.
22
Study schematic. Timing of visits and data collection. Note: The following data will also be collected: Date of discontinuation of ECCO2R and reason; Date of discontinuation of mechanical ventilation (unassisted breathing); Date of critical care discharge; Date of hospital discharge and Date of death.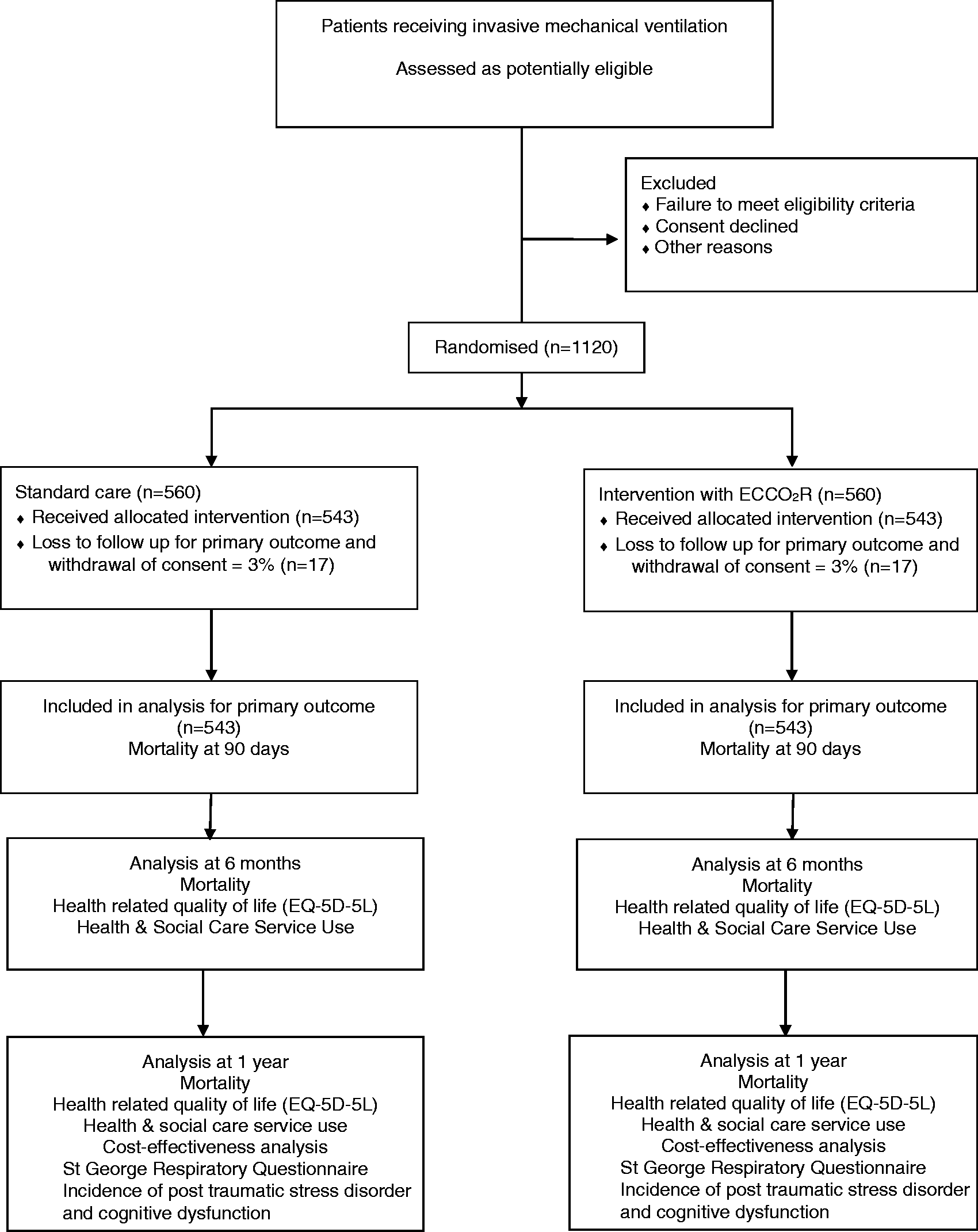

Statistical analysis
The primary analysis will be conducted on all outcome data obtained from all participants as randomised and regardless of protocol adherence, i.e. intention to treat analysis.
For the primary outcome and other dichotomous outcomes, risk ratios and 95% confidence interval (CI) will be calculated. The primary outcome of 90-day mortality will be analysed using chi-square and a secondary analysis using logistic regression to adjust for important covariates will also be carried out. The comparison of continuous outcomes between the two groups will be investigated using analysis of covariance, adjusting for other covariates where appropriate. Time-to-event outcomes will be analysed by survival methods and reported as hazard ratios with 95% CI. The intention-to-treat basis analysis will use a significance level of less than 0.05. Sensitivity analysis will be performed for the primary outcome, excluding the first two intervention arm patients at each site in order to address potential learning effects. This will be explored further using a model-based approach. A detailed statistical analysis plan will be developed during the trial, submitted to the DMEC for review and made publically available prior to the commencement of analysis.
An independent statistician on the DMEC will conduct an interim analysis for the primary outcome measure, before the recruitment of 560 patients. Using the chi-square statistic (mortality by treatment group), a p-value less than 0.001 will be used according to the Haybittle-Peto stopping rule.
Missing data
Every effort will be made to minimise missing baseline and outcome data in this trial. The level and pattern of the missing data in the baseline variables and outcomes will be established by forming appropriate tables and the likely causes of any missing data will be investigated. This information will be used to determine whether the level and type of missing data has the potential to introduce bias into the analysis for the proposed statistical methods, or substantially reduce the precision of estimates related to treatment effects. If necessary, these issues will be dealt with using multiple imputation or Bayesian methods for missing data as appropriate.
Health economics evaluation
A cost-effectiveness analysis will be undertaken to compare the costs and outcomes associated with vv-ECCO2R to those associated with standard care at two time points. The initial, within-trial analysis will be based on the costs and outcomes measured within the one-year study period, while the second model-based analysis will extrapolate the cost and outcomes over a lifetime horizon. Both analyses will estimate the cost per quality adjusted life year (QALY) gained. They will adhere to the National Institute for Health and Care Excellence’s (NICE) guide to methods of technology appraisal, 23 where appropriate. The analyses will be performed from the perspective of the National Health Service (NHS) and personal social services (PSS). For the within-trial analysis, patients’ use of health and social care resources over the study period will be collected from baseline up to one year. This will include duration and level of critical care received and hospital ward length of stay, therapeutic procedures received (including transfusions), subsequent outpatient visits and patient contacts with primary care. Resource use associated with the primary admission will be collected using the case report form. Health and social care service use following discharge to 12 months will be captured via self-completed resource questionnaires at 6 and 12 months. Log sheets will be provided at discharge and six months to help patients keep track of their service use. Costs will be calculated by attaching appropriate unit cost from publicly available sources (e.g. Department of Health National Schedule of Reference Costs). Utilities for the calculation of the QALYs will be gathered using the EQ-5D-5L administered at six months and one year. 24 These will be completed by the patients where possible and by a proxy where not.
Standard methods will be used to explore and display uncertainty in the cost-effectiveness data including scatterplots on the cost-effectiveness plane and cost-effectiveness acceptability curves (CEACs). Sensitivity analysis will be performed to assess the robustness of the cost-effectiveness results to changes in key parameters (including the cost of the vv-ECCO2R device). A decision-analytic model will be developed in order to estimate the lifetime cost-effectiveness. The model will be populated by both primary cost and outcome data derived from the within-trial analysis and data extracted from secondary sources. The model structure will be informed by a review of the literature and will follow accepted guidelines for good practice in decision-analytic modeling.23,25 Probabilistic sensitivity analyses will be undertaken to investigate uncertainty surrounding the estimates of lifetime costs.
Data monitoring
The NICTU will be responsible for trial monitoring and visits will be conducted in accordance with the trial monitoring plan and will comply with the principles of Good Clinical Practice (GCP). On-site monitoring visits during the trial will check the accuracy of data entered into the clinical trial database against the source documents, adherence to the protocol, procedures and GCP, and the progress of patient recruitment and follow-up.
Safety, ethics and dissemination
Adverse outcomes
The European Commission guidance document “Guidelines on Medical Devices – Clinical Investigations – Serious Adverse Event Reporting” MEDDEV 2.7/3 provides the definitions for the adverse events. This is based on the International Organisation for Standardisation (ISO) published revised standard on good clinical practice requirements for medtech clinical investigations, ISO 14155:2011 “Clinical Investigation of medical devices for human subjects” – Good clinical practice. The word “event” is used for untoward medical occurrences not related to the investigational device. The word “effect” is used for occurrences related to or caused by the investigational device.
List of expected adverse events.

Adverse events will be graded according to severity, seriousness, causality and expectedness. All those adverse events meeting the definition as serious will be submitted to the clinical trials units within 24 h of the investigator becoming aware.
Regulatory and ethical approvals
This is an investigator-led study of a marketed medical device for which there is a labeled indication. As the trial will be employing a medical device for a purpose for which it has approval, and the device has a CE mark, approval from the Medicines and Healthcare products Regulatory Agency (MHRA) will not be required.
Ethical approval from a multicentre research ethics committee (MREC) flagged for trials involving patients without capacity has been obtained for England, Wales and Northern Ireland (16/SC/0089) and Scotland (16/SS/0048).
The study will comply with the principles for sharing clinical trial data from publicly funded clinical trials.
Confidentiality
In order to maintain confidentiality, all CRFs, questionnaires, study reports and communication regarding the study will identify the patients by the assigned unique trial identifier and initials only. Patient confidentiality will be maintained at every stage and will not be made publicly available to the extent permitted by the applicable laws and regulations.
Dissemination
The study will be reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) guidelines and the template for intervention description and replication (TIDieR) checklist and guide.26,27
The study findings will be presented at national and international meetings with abstracts on-line. Presentation at these meetings will ensure that results and any implications quickly reach all of the UK intensive care community. In accordance with the open access policies proposed by the NIHR, we aim to publish the clinical findings of the trial as well as a paper describing the cost-effectiveness in the NHS setting in high quality peer-reviewed open access (via Pubmed) journals. The NIHR HTA requires that a detailed study report is published in the HTA journal.
An on-going update of the trial will also be provided on the NICTU website (http://www.nictu.hscni.net/rest-trial/) and twitter (@resttrial).
Discussion
The REST study aims to determine the clinical and cost-effectiveness of vv-ECCO2R and lower tidal volume ventilation in patients receiving mechanical ventilation for acute hypoxaemic respiratory failure. This will be the largest trial evaluating vv-ECCO2R and lower tidal volume mechanical ventilation to date.
The study population is defined by hypoxaemia as opposed to ARDS. This decision was informed by evidence that the benefits of lower tidal volume mechanical ventilation also apply to those patients without ARDS 28 and this was reflected in the HTA commissioning brief which indicated the study population should be adult patients with moderately severe acute hypoxaemic respiratory failure, rather than ARDS. The study does not therefore require the interpretation of chest radiographs to enable the diagnosis of ARDS, which is prone to inter-observer variability. 29
This is an open and pragmatic trial in which blinding to the intervention is challenging due to its invasive nature. This potential bias is the main potential limitation of the trial. To overcome this, the objective outcome of 90-day mortality was used as the primary outcome measure. It is recognised that in an unblinded study in this population if an earlier time point such as 28-day mortality is used, it is possible that bias related to withdrawal of care due to futility in ICU might be a risk. By using 90-day mortality, this is avoided as decisions to withdraw care due to futility in ICU are typically made much earlier than day 90. Furthermore, in support of using 90-day mortality as the primary outcome measure, it is increasingly recognised that 28-day mortality does not capture all of the acute mortality related to ARDS and that mortality from ARDS does not stabilise to days 60–90.
Another potential limitation relates to the necessary use of heparin anticoagulation to enable vv-ECCO2R in the intervention arm as a possible confounding therapy which might independently be associated with improved outcomes in ARDS and sepsis, although this is unproven. 30 Finally, the potential for increased requirement of positive end expiratory pressure in the intervention arm as a result of tidal derecruitment associated with lower tidal volume ventilation may also independently affect patient outcomes. 31 These potential confounders reflect the complex nature of undertaking trials in this study population. Regardless, if effective, we will be able to conclude that this package of care was able to achieve the planned outcome.
A recent pilot study of lower tidal volume ventilation facilitated by vv-ECCO2R confirmed the feasibility of our planned intervention. 32 By definition, a pragmatic study aims to assess the intervention delivered in the setting of standard care, compared to standard care alone. In order to help ensure compliance with standard care, we will audit ventilator settings every three months and give feedback to sites regarding their compliance with best practice. Although there are adjunctive measures used in the management of hypoxaemic respiratory failure which may be beneficial, such as neuromuscular blocking drugs and prone positioning, these measures are not used routinely in our patient cohort. 21 We have therefore not mandated their use in either arm of the study.
There are several vv-ECCO2R devices available. A single device was used to avoid the necessity for participating sites to train on multiple devices. The Alung device was used specifically due to its relative ease of use and lower extra-corporeal blood flows which are comparable to blood flows used with continuous renal replacement therapy with which all ICUs in the study will be familiar. As the trial aims to evaluate lower tidal volume ventilation, the results should be generalisable to any vv-ECCO2R device which can facilitate lower tidal volume mechanical ventilation.
In conclusion, if this trial finds that lower tidal volume ventilation results in improved outcomes and is cost effective, then this has the potential to change clinical practice in terms of how we manage patients with acute hypoxic respiratory failure. Moreover, if it is found that its use does not result in improved outcomes, then this will be important to avoid its use in these circumstances and prevent an ineffective technology becoming embedded in clinical practice.
Footnotes
Authors’ contributions
JMN and DFM conceived the study. All authors made a substantial contribution to the protocol development. CMD is the study statistician, JMN and DFM wrote the first draft of the manuscript and all authors critically revised the manuscript and gave final approval of the version to be published. JMN and DFM are accountable for all aspects of the work. The support of the UK INtensive Care Foundation is gratefully acknowledged. The authors acknowledge the support of the Northern Ireland Clinical Rersearch Network and the National Institute for Health Research Critical Care Research Network for this study.
Acknowledgements
DFM, GDP and AJG are Co-Directors of Research for the Intensive Care Foundation. We thank all staff of the Northern Ireland Clinical Trials Unit for their support (C McNally, C Jackson, R Boyle, P Frew, M Guiney, G Harkness, J Johnston, G Phair, P Rafferty, S Toase, M Wilson). We wish to thank M Campbell, T Clutton-Brock, N Ferguson and B Taylor Thompson as members of the data monitoring and ethics committee and W McGuire, B Williams and A Nichol as members of the trial steering committee.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: DFM reports grants from the NIHR HTA for the conduct of the study. In addition, DFM reports personal fees from consultancy from GlaxoSmithKline, Bayer, SOBI and Peptinnovate. His institution has received grants from NIHR and others as well as funds from GlaxoSmithKline for DFM undertaking bronchoscopy as part of a clinical trial. DFM also holds a patent for a treatment for ARDS issued to Queen’s University Belfast. NAB has received educational and research funding from Maquet, Drager, Fisher & Paykel, Mitsubishi Tanabe Pharmaceuticals, Corpak, ALung Technologies. DB has received educational funding from Alung Technologies and Kadence. GDP reports personal fees from GlaxoSmithKline.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research grant has been awarded by the National Institute for Health Research Health Technology Assessment (HTA) programme [13/143/02]. SJB and ACG are supported by the NIHR Comprehensive Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London. MG is funded by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. GDP is supported as a NIHR Senior Investigator. The views expressed in this publication are those of the authors and not necessarily those of the NHS, The National Institute for Health Research or the Department of Health.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.