
Abstract
The main objective of molecular docking is to find a model of interaction between a protein and ligand with a minimum binding energy. This process is driven by intricate algorithms and scoring functions. This paper mainly concentrates on the search algorithm used for solving the docking problem. Here, a new approach is proposed for the molecular docking problem that utilizes a hybrid algorithm that combines an improved quantum-behaved particle swarm optimization algorithm (QPSO) and the Solis and Wets algorithm. The improved QPSO algorithm that is based on individual particle evolutionary processes is known as individual particle evolutionary particle swarm optimization (IEQPSO). The IEQPSO algorithm was tested and compared with particle swarm optimization, QPSO, and its variants with a suite of benchmark functions. The results indicated the superiority of the proposed approach according to benchmark test functions. Then, the hybrid algorithm based on the IEQPSO algorithm was used for optimizing the energy function of the molecular docking problem and was compared with the classical Lamarckian genetic algorithm used by molecular docking software. Molecular docking and molecular dynamics simulation experiments revealed the effectiveness and feasibility of the proposed algorithm in solving the molecular docking problem.
Keywords
Introduction
Among many evolutionary algorithms, the quantum-behaved particle swarm optimization (QPSO) algorithm 1 is more efficient than other conventional algorithms. It is based on quantum mechanics and trajectory analysis of particle swarm optimization (PSO). 2 The main difference between QPSO and PSO is that the QPSO algorithm requires no velocity vectors for particles, and it has fewer parameters to adjust. The QPSO algorithm has been shown to successfully solve a wide range of optimization problems. Moreover, many efficient strategies have also been proposed to improve algorithm performance.3–4 However, in the QPSO algorithm, the mean best position is simply the average of the personal best position of all particles, which means that each particle is considered equal and exerts the same influence on the mean best position. In fact, it is not appropriate to consider each member equal. In this article, to improve the performance of QPSO in highly dimensional complex problems, we proposed an improved QPSO based on an individual particle evolutionary process (individual particle evolutionary quantum-behaved particle swarm optimization (IEQPSO)) algorithm to improve the global search capability and the convergence performance of the optimization process. The benchmark functions were used to test the performance of the IEQPSO algorithm. The ultimate aim of the optimization algorithm is to solve real-world problems. Here, we use the IEQPSO algorithm to optimize the energy function used for the molecular docking problem.
Molecular docking is an important tool in drug discovery, and many docking methods have been developed and widely used. 5 It can be used for predicting the correct conformation between a protein and ligand, for identifying novel lead compounds, for preliminary lead optimization, 6 and for investigating the mechanisms of action of biologically active compounds. 7 The quality of molecular docking depends on two factors: the scoring function and the search algorithm. 8 The scoring function aims to evaluate the conformations between a protein and a ligand to detect those conformations with the minimum binding energy. The search algorithm is used to identify the most favorable ligand conformation when the ligand binds to the receptor protein.
Various optimization algorithms and docking software have been proposed for use in solving different aspects of molecular docking problems. For the docking software, a popular tool used for molecular docking is AutoDock, 9 which uses the C++ software package. In addition, there are many other excellent software programs, such as DOCK, 10 GOLD, 11 FlexX, 12 Glide, 13 and PythDock, 14 and so on. With the development of docking software, a number of different metaheuristics have been proposed as search methods to solve the docking problem. Although all the above mentioned works are helping in solving molecular docking problems, due to the complexity of docking free energy landscapes with rugged funnel shapes, molecular docking is still a very challenging problem.
Based on above description, the paper highlights the following: (1) We proposed a new variant version of the QPSO algorithm, called the IEQPSO algorithm. The modification involves the use of the individual particle evolutionary process to calculate the mean best position to strike a balance between the needs of the global search process and the local search (LS) process. (2) To solve the molecular docking problem, a hybrid strategy based on combining the IEQPSO algorithm with the Solis and Wets algorithm 15 was proposed for dealing with the energy optimization problem. (3) The protein 1B58 16 was used as an experimental receptor model, and the detailed mechanisms involved in the protein–ligand binding interactions of the compounds were elucidated from the results of the molecular docking and molecular dynamics (MD) simulations.
The remaining exposition is as follows. The upcoming section introduces the related state-of-the-art stochastic optimization methods for molecular docking problems. Then, a subsequent section describes the proposed method. The results are discussed in penultimate section, and the final section concludes the paper.
Related state-of-the-art stochastic optimization methods
Many researchers have proposed various methods for predicting molecular docking interactions. The search algorithms mainly include simulated annealing (SA), Monte Carlo (MC), genetic algorithm (GA), PSO, differential evolution (DE), and so on, and the many variants of these algorithms. For example, the GA and SA were first used in AutoDock software, and Morris et al. 9 proposed the Lamarckian genetic algorithm (LGA), which is a hybrid of GA and LS, to minimize the binding energy. The LGA search method is more efficient in escaping from a local minimum. In addition, there are many other remarkable studies that use GA to solve protein–ligand problems. Kang et al. 17 presented an improved adaptive GA to solve the problem of protein–ligand docking. Li et al. 18 used an entropy-based GA with multi-population evolution to solve the docking problem and created a coefficient adaptive scoring function that greatly improves the docking accuracy. Guan et al. 19 presented a novel and robust optimization algorithm based on the LGA. This algorithm applied a population evolution direction-guided model of genetics and could find solutions with lower energy for flexible protein–ligand docking problems. Leonhart et al. 20 developed the Biased Random Key Genetic Algorithm (BRKGA-DOCK) as a sampling strategy for the protein–ligand docking problem. The proposed method has been compared to other existing tools and obtains the best average root-mean-square deviation (RMSD). Guan et al. 21 proposed a novel GA based on a crossover elitist preservation mechanism (crossover elitist preservation genetic algorithm mechanism (CEPGA)) for solving protein–ligand docking problems. The algorithm employed crossover elitist preservation to keep the elite individuals of the last generation and make the crossover more efficient and robust. The algorithm was compared with GA, LGA, and SODOCK, and the results indicated that CEPGA could obtain the lowest energy and the highest accuracy.
The MC algorithm has also been studied for the molecular docking problem. For example, in the study by Liu and Wang, 22 an MC simulation approach was used for instances of flexible ligands. Lee et al. 23 applied conformational space annealing (CSA) and the MC algorithm with the minimization (MCM) method to generate global optimization methods to perform molecular docking simulations. The simulation results showed that the CSA method is more efficient and accurate than the MCM method.
The DE algorithm is an alternative for the molecular docking problem. Koohi-Moghadam and Rahmani 24 used an enhanced DE algorithm with an LS algorithm and a pseudo-elitism operator to address the docking problem. Guan et al. 25 introduced an efficient ABC_DE-based hybrid algorithm for protein–ligand docking, integrating artificial bee colony algorithm and DE algorithm.
More recently, Swarm Intelligence techniques were used to tackle molecular docking problems. For example, Liu et al. 26 introduced a fully informed swarm optimization algorithm (FIPSDock) for solving flexible protein–ligand docking problems. The García-Nieto et al. 27 utilized multiobjective particle swarm optimization (MPSO) variants based on different archiving and leader selection strategies to minimize the intermolecular energy and the RMSD in the predicted ligand conformations. In the study by Dai et al., 28 a new MPSO algorithm based on decomposition (MPSO/D) was proposed for the molecular docking problem. The results showed that the proposed algorithm had good performance in terms of convergence and diversity. García-Nieto et al. 29 proposed a new variant of speed-constrained multi-objective PSO (SMPSO) algorithm, which is characterized by the use of archive strategies. In the study, the authors analyzed several SMPSO variants based on different archiving strategies for molecular docking.
Based on reports in the literature, we have observed that GA-based methods have been prominent when solving the molecular docking problem. At the same time, PSO methods have been applied to these problems more frequently in recent years.
Methods
Quantum-behaved particle swarm optimization
We assume that during QPSO with M individuals, at the nth iteration, particle i flies in the N-dimensional quantum space.
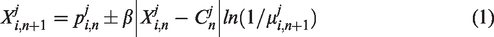
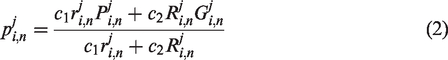
The parameter β is the contraction-expansion (CE) coefficient.
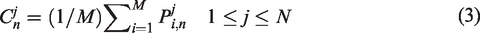
Individual particle evolutionary QPSO algorithm
In QPSO, the mean best position is introduced to make the algorithm more efficient than the original algorithm. By using equation (3), the mean best position is calculated using the average of the personal best position of all particles; however, the effect and influence of each particle are not exactly the same. Considering the characteristics of each particle in the evolutionary process, we designed a new control method to replace the mean best position in the QPSO in this section. A new parameter was introduced, which is based on the ratio between the fitness value of the global best solution

Then, by introducing the new parameter control, the mean best position equation is rewritten as follows

In equation (5), the parameter
Pseudocode of IEQPSO:

Results and analysis
Performance evaluation and comparison of benchmark functions
To evaluate the overall performance of the proposed IEQPSO during function optimization, the algorithm was tested with eight well-known benchmark functions (Table 1). For the performance comparison, PSO with inertia weight (PSO-In), the original QPSO algorithm and quantum-behaved particle swarm optimization with weighted mean best position (WQPSO) 30 were also tested with the benchmark functions. Parameter settings for these algorithms are listed in Table 2. The mean best fitness value and standard deviation based on 50 runs of each algorithm on each problem are presented in Table 3. The standard deviation values are shown in brackets.
Benchmark functions.

Parameter settings for the PSO-In, QPSO, WQPSO and IEQPSO algorithms.

PSO: particle swarm optimization; QPSO: quantum-behaved particle swarm optimization; WQPSO: QPSO with weighted mean best position; IEQPSO: individual particle evolutionary QPSO; CE: contraction-expansion.
Results of the benchmark functions.

PSO: particle swarm optimization with inertia weight; QPSO: quantum-behaved particle swarm optimization; WQPSO: QPSO with weighted mean best position; IEQPSO: individual particle evolutionary QPSO. The optimal solution of each problem is presented in boldface in Table 3 for four algorithms.
For the shifted Sphere Function (f1), all the algorithms could obtain a solution near the global optimal solution, but the IEQPSO algorithm obtained better results than the other methods. The results for the shifted Schwefel’s 222 function (f2) show that IEQPSO could find a global optimum successfully and exhibited a better search ability than PSO-In and QPSO. For the DeJong function (f3), the WQPSO algorithm outperformed the other methods. In regard to benchmark f4, the Rosenbrock function is a multimodal function, which is too difficult to solve for many optimization problems. The IEQPSO algorithm showed a slightly better performance among all the algorithms, and its obvious advantage over other algorithms is shown by its standard deviation values. The fifth benchmark function, f5, is the Ackley function, and the results indicate that all the QPSO-based methods showed better performance for this problem than PSO. For function f6, the Rastrigrin function, the IEQPSO algorithm yielded the best result. For this function, the difference between QPSO and PSO-In was not significant. Based on the results obtained for f7, the Schaffer problem, IEQPSO had a significant advantage over the other methods. The results for the shifted, rotated Griewank’s function (f8) suggest that the IEQPSO algorithm was able to find a better solution than the others.
Figure 1 presents the convergence process of the algorithms for each benchmark function. It shows that the IEQPSO algorithm had a better convergence property than its competitors during the later stage of iteration, except for functions f3 and f5. According to the results shown above in the tables and figures, it can be seen that the IEQPSO algorithm was able to find solutions of higher quality for the functions compared to those obtained using the other methods, in most cases.

Convergence process of the algorithms for each benchmark function.
Results for molecular docking
Because the energy search during molecular docking is complex, we used a hybrid strategy based on combining the IEQPSO algorithm with the Solis and Wets algorithm to address energy optimization that was referred to as the hybrid algorithm of IEQPSO and local search (LIEQPSO). The IEQPSO algorithm is used as a global search algorithm, and the Solis and Wets algorithm is used as an LS algorithm. The addition of an LS algorithm effectively maintains the diversity of the particles and prevents premature convergence of the algorithm. In addition, the LGA algorithm was used as the other search algorithm. It is the classical algorithm used in AutoDock 4, in which the GA algorithm incorporates the Solis and Wets algorithm during the LS. The main motivation for this is to analyze the docking performance of both algorithms to determine which of them can obtain better scoring values. To achieve this goal, we attempted to incorporate the new algorithm into the source code of AutoDock 4.
Problem description
AutoDock 4 software is used to evaluate a docked conformation. The scoring function of AutoDock 4 is the semiempirical free energy function. The total docked energy is expressed as the sum of the intramolecular energy and intermolecular energy of the protein and ligand. The intramolecular energy was estimated for the transition from the unbound state to the bound state of the protein and ligand. The intermolecular energy was estimated for the combination of the protein and the ligand in the bound conformation.
The force field includes six pairwise estimates (V) and the evaluation of the conformational entropy lost upon binding (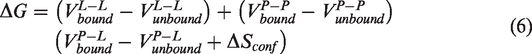
Each of the pairwise energetic items consists of the following sources of energy: the first source is the Lennard-Jones 12–6 Van der Waals interaction energy, the second source is the 12–10 hydrogen bond potential energy, the third source is the electrostatic potential energy, and the final is the desolvation potential energy.

Experimental results for protein–ligand docking
The protein and ligand docking tests were run on the Red Hat Enterprise Linux OS. The prepared protein and ligand structures were saved in the PDBQT file format. The AutoDock package includes two programs, the AutoGrid program, and the AutoDock program. The AutoGrid program performs the calculation of the energy grid maps. The grid size was set to 60 × 60 × 60 points with a spacing of 0.375 Å. The AutoDock program is responsible for the conformational search and energy evaluation. Table 4 shows the parameter settings of the search algorithms used for docking experiments. The other parameters used the default settings in AutoDock 4.
Parameter settings for protein–ligand docking.

For the test case, PDB 1B58 was used as the experimental receptor protein and was obtained from the RSCB protein data bank (http://www.rcsb.org/). The sequence Lys-Tyr-Lys was used as the experimental ligand and contained 43 atoms.
There are two criteria for evaluating molecular docking results: the binding energy and RMSD of docking. The RMSD represents the comparison between atomic positions in a docked ligand and the corresponding positions in the ligand crystal structure. In general, we assumed that the lower the binding energy, the closer the docked complex was to the native state of the complex. The lower RMSD value is assumed to have higher docking accuracy. An RMSD value of 2 Å was usually used as the cutoff value. For the LIEQPSO docking complex, the best binding result had an energy = −19.68 kcal/mol and an RMSD = 1.63 Å. The best result for the LGA docking complex had an energy = −12.74 kcal/mol and an RMSD = 2.00 Å. According to the lowest energy and RMSD values, the LIEQPSO algorithm obtained a better docking result than the LGA algorithm.
Among the sources of energy involved in docking binding, Van der Waals energy and electrostatic interaction energy are the principal binding forces in protein–ligand interactions. For Van der Waals energy, both attractive and repulsive interactions play a vital role in controlling the binding phenomenon. The electrostatic interaction plays an important role in changing the enthalpy resulting from binding. The Van der Waals interaction energy and the electrostatic binding energy of the ligand residue atoms are plotted in Figure 2. A more negative value for binding energy indicates favorable binding of ligand with the protein receptor. Figure 3 shows examples of complex docking obtained by the two methods. Based on the above results, the LIEQPSO algorithm has a higher docking accuracy than the LGA algorithm for the test case.

Van der Waals interaction and electrostatic interaction energy calculated for the ligand residue atoms of the complexes docked by the LIEQPSO and LGA algorithms.
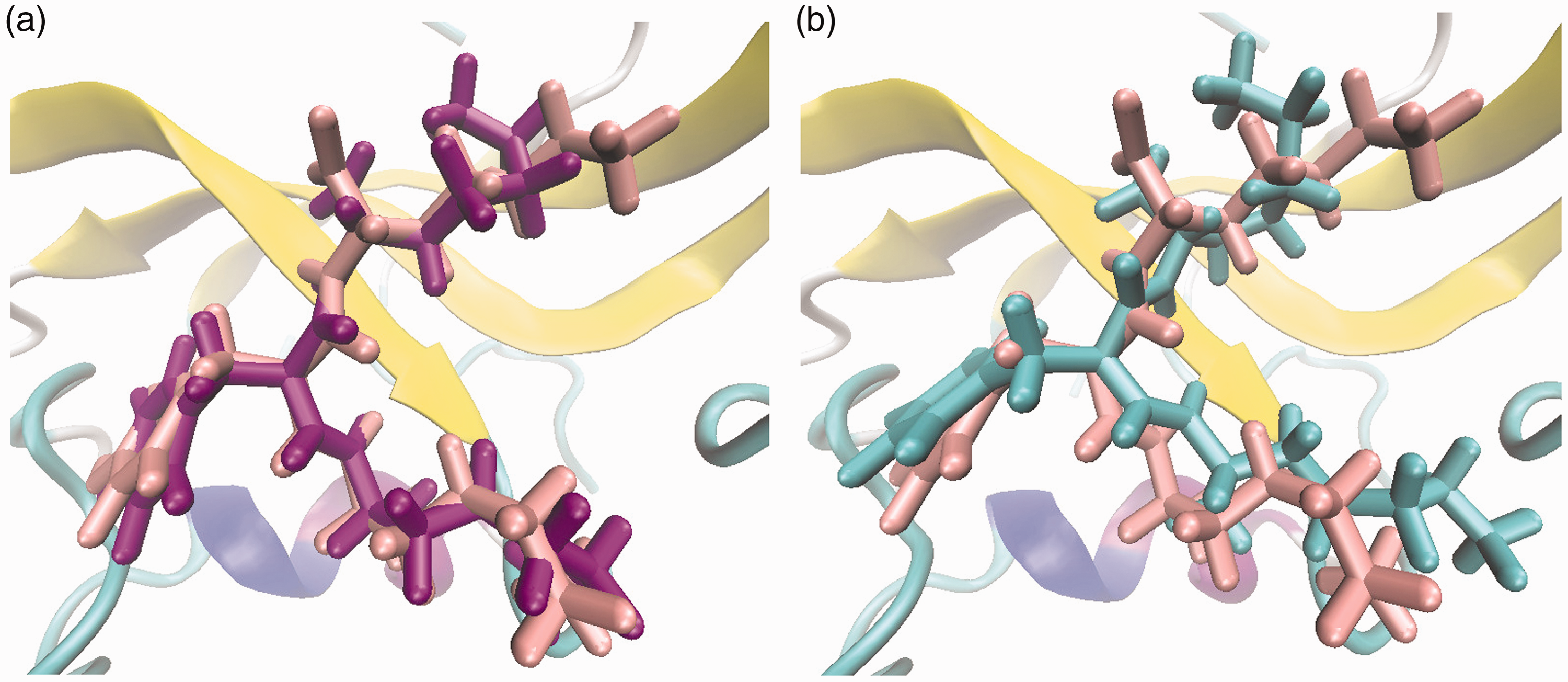
Examples of complex docking obtained by the (a) LIEQPSO algorithm and (b) LGA algorithm. The receptor protein is represented by a novel cartoon secondary structure style. The color of the crystal structure is pink, and the predicted structures are purple (a) and cyan (b).
MD simulation results for protein–ligand docking
The objective of docking is to obtain a model of the interaction between a ligand and a protein that is characterized by a minimum binding energy. For further detailed analysis of protein–ligand interactions, we performed MD simulations with the docking results to establish a more reliable method to illustrate protein–ligand interactions. The interaction of proteins with ligands is very important in biological processes in the context of drug design and pharmacology, since proteins usually accomplish their functions through binding to various molecules. In this section, we used NAMD software 31 and the CHARMM27 force field 32 to perform the MD simulations. The simulated temperature was set to 300 K, and the simulation time was set to 10 ns.
Complex stability analysis
To evaluate the overall structural stability and flexibility of the complex structure, we calculated the RMSD of the complex structure. The RMSD is defined as follows.
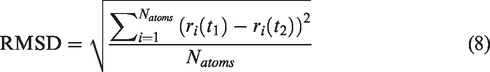
The RMSD of the backbone atoms in the complex versus the simulation trajectory for 10 ns was calculated. As shown in Figure 4(a), we noticed that the RMSD of the LIEQPSO docking complex structure fluctuated around a value of 2.5 Å after 6 ns. The average RMSD value is 2.05 Å for the backbone atoms. In the MD simulation of the LGA docked complex, the backbone RMSD fluctuated between 0.49 Å and 2.78 Å during the simulation. The average RMSD value is 1.99 Å for the LGA docked complex. The plots showing the RMSD of the backbone atoms could indicate the stability of the complexes in the MD simulation.
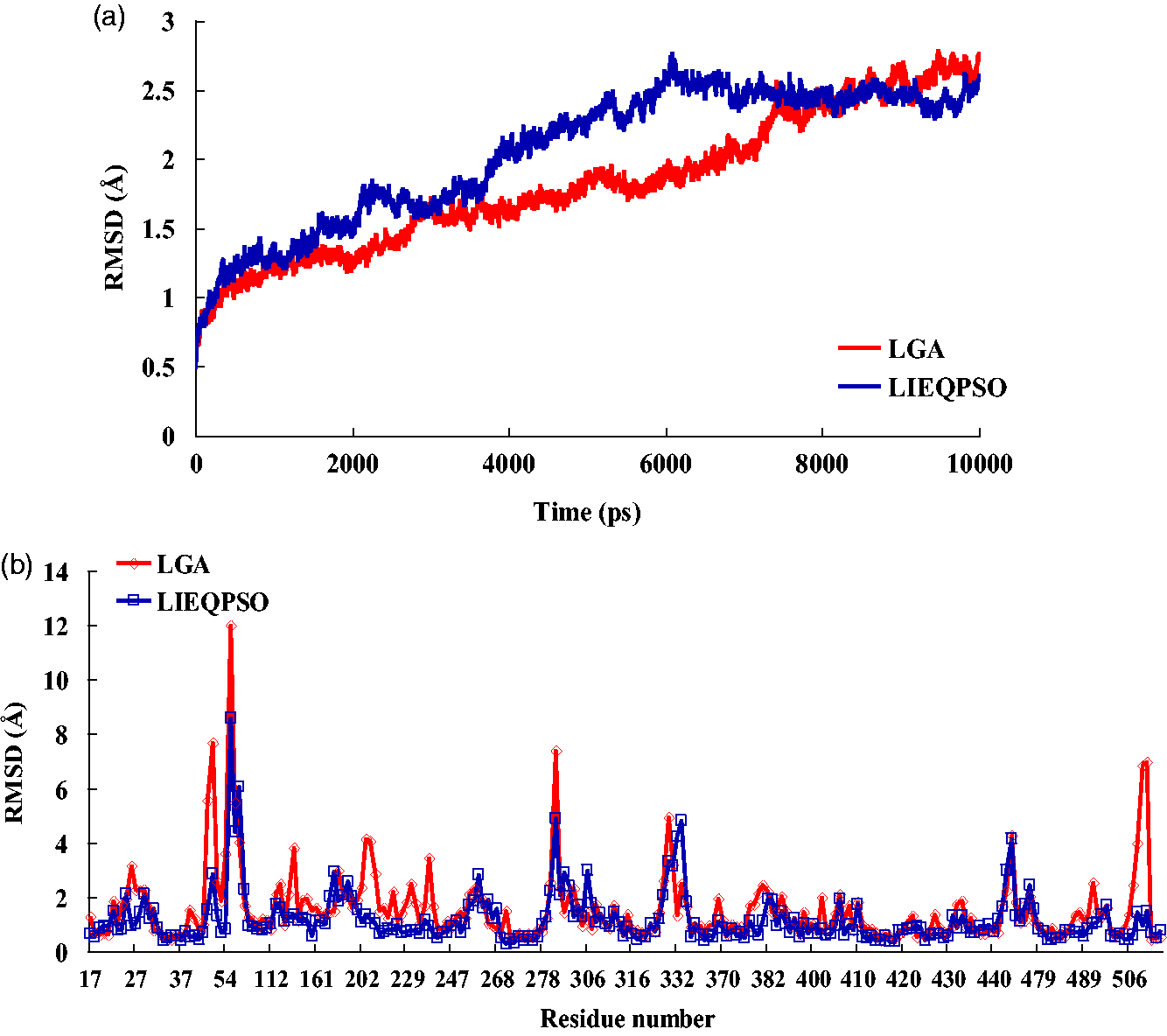
(a) RMSD of the backbone atoms as a function of time in the docked complex and (b) RMSD as a function of the residue number in the docked complex at a temperature of 300 K.
To investigate whether there are any specific dynamic residues involved in the protein–ligand interaction, the RMSD of the residues was calculated. This can indicate the flexibility of the protein residues. It can also clearly represent the differences in some residues in both complexes. As shown in Figure 4(b), a comparison of the RMSD of the structures of the LIEQPSO complex and the LGA complex revealed that the RMSD curves exhibited great similarity in their fluctuation trends, with only a few residues showing a significant difference. For instance, the RMSD values of residue PHE44 according to the LGA docking results were 7.68 Å, whereas the RMSD was 2.88 Å in the LIEQPSO docking complex. Similar residues, including PRO56, VAL221, and LEU512, among others, also showed higher RMSD values in the LGA docking complex. In general, the flexibility of the residues in the LIEQPSO docking complex was relatively lower than those in the LGA docking complex.
Salt bridge between interfacial residues
In the interactions between proteins and ligands, electrostatic interactions play an important role due to their high specificity. We used a salt bridge to examine the contribution of electrostatic interactions. Here, we have only considered the salt bridge present at the interface of the complex. A salt bridge was identified according to a cutoff distance of 4.0 Å between any of the nitrogen atoms in the basic residues and the oxygen atoms in acidic residues. It was found that there were two salt bridges in the interfacial residues in the complex formed by docking by the LIEQPSO algorithm at 300 K (Figure 5(a)). LYS1 of the ligand forms a salt bridge with the ASP419 residue of the protein, and LYS3 of the ligand forms a salt bridge with the GLU229 residue of the protein. In the complex that was docked by the LGA algorithm, one salt bridge was found, and the LYS1 residues in the ligand formed a salt bridge with ASP419 in the protein (Figure 5(b)). As shown in Figure 5, the salt bridge LYS1-ASP419 in the complex docked by the LIEQPSO algorithm was not as stable as that in the complex docked by the LGA algorithm. The salt bridge ASP419-LYS1 in the LGA docked complex was mostly maintained at a distance of 4.0 Å. In comparison, the salt bridge ASP419-LYS1 in the LIEQPSO docked complex was ruptured and restored throughout the simulation process. Fortunately, the two salt bridges that existed in the complex docked by the LIEQPSO algorithm were believed to promote the stability of the complex.
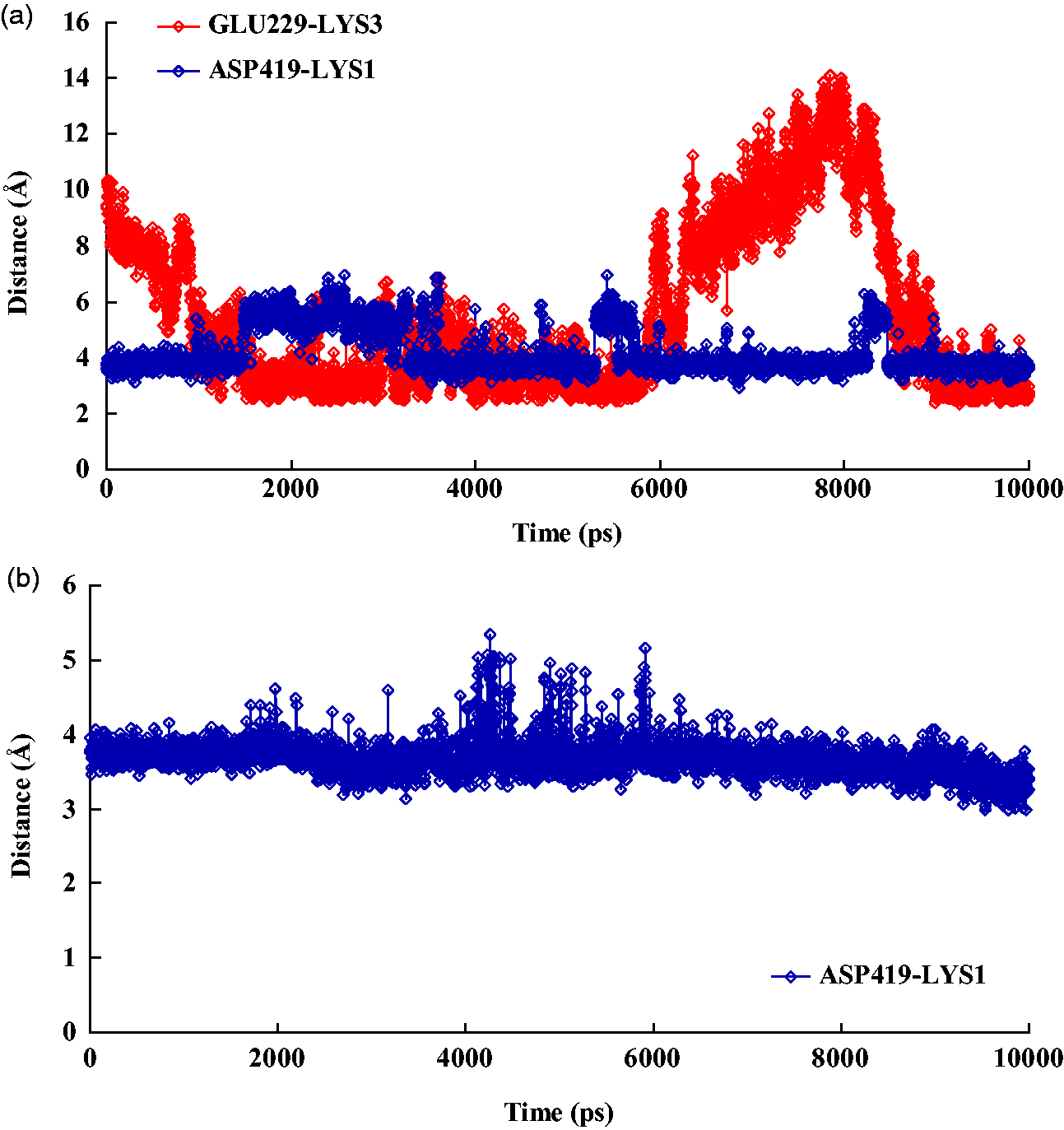
Plots showing changes in the salt bridge in the binding domain as a function of time. (a) Changes in the salt bridges ASP419-LYS1 and GLU229-LYS3 in the LIEQPSO docking complex and (b) changes in the salt bridge ASP419-LYS1 in the LGA docking complex.
Hydrogen bond in between interfacial residues
Hydrogen bonds are also important factors that influence the binding stability of protein ligands. The presence of hydrogen bonds is an important indication of the significance of protein–ligand interactions. In the complex, interfacial residues forming hydrogen bonds are shown in Figure 6. For the complexes docked by both algorithms, hydrogen bond residues within the protein are mainly concentrated near ARG413, CYS417, and VAL34. The obvious distinction is that the hydrogen bonds of both complexes have different occupancy times.

Hydrogen bond map between protein and ligand interfacial residues in the complex.
For the complex after molecular simulation, a distance cutoff of 3.5 Å and an angle cutoff of 30° were used for the hydrogen bond calculation. According to the above mentioned hydrogen bond calculation criteria, we have listed the hydrogen bonds in interfacial residues that exist >50% of the time throughout the entire simulated trajectory at 300 K. We observed 10 hydrogen bonds in the LIEQPSO docked complex and 10 hydrogen bonds in the LGA docked complex. Among these hydrogen bonds, seven common hydrogen bonds, ARG413::LYS3, CYS417::LYS1, ASP419::LYS1, GLU32::TYR2, VAL34::TYR2, ARG404::TYR2, and LYS1::HSD161, were found in both complexes. Although ALA418::LYS1, ASN247::LYS3, and ASN246::LYS3 were also found in the LGA docked complex, they exist for a relatively short time during the MD trajectory. The results show that three hydrogen bonds, ARG413::LYS3, CYS417::LYS1, and ASP419::LYS1, were the most stable in both complexes. In general, the hydrogen bonds in the LIEQPSO docked complex have higher occupancy time in the simulated trajectory (Table 5).
The major hydrogen bonds in the binding domains of both complexes.

LGA: Lamarckian genetic algorithm; LIEQPSO: hybrid algorithm of IEQPSO and local search.
Conclusion
This paper describes an improved QPSO algorithm based on individual particle evolutionary processes. A hybrid algorithm of IEQPSO and the Solis and Wets method was used to study molecular docking problems. Finally, we presented an MD simulation investigation on the binding interactions between proteins and ligands.
To assess the performance of the IEQPSO algorithm, eight test cases with unimodal functions and multimodal functions were used for comparison. The results indicated that the IEQPSO algorithm significantly improved the search ability and convergence precision compared to other algorithms.
To solve the molecular docking problem, the ligand can be bound to the protein by the LIEQPSO and LGA algorithms. The MD simulation results indicated that there were complex binding interactions between the protein and the ligand. The results indicate that the proposed LIEQPSO algorithm outperforms the LGA algorithm in terms of optimal solutions. Based on the above analysis, the proposed LIEQPSO algorithm may be used as an alternative method for solving molecular docking problems.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the National Natural Science Foundation of China under Grant Nos. 61502203, the Natural Science Foundation of Jiangsu Province under Grant Nos. BK20150122, the Natural Science Foundation of the Jiangsu Higher Education Institutions of China under Grant Nos. 17KJB520039 and Nos. 18KJB520046, the 333 High-level Talents Training Project of Jiangsu Province under Grant Nos. BRA2018147, “Qing Lan Project” of Wuxi City College of Vocational Technology.