
Abstract
This study systematically investigated the influence of substituents (NH2, CH3, CF3, and Cl) on the keto-enol tautomerism of 1,2-cyclodiones, focusing on reactivity and thermodynamic stability. Calculations were performed using the M06-2X/6-311G++** level of theory, with a solvation model through CPCM/M06-2X/6-311G++**. Results indicate ring-size dependent tautomerism, with three-, five-, and six-membered rings favouring the keto-enol form, while four- and seven-membered rings favour the diketo form. The NH2 substituent uniquely stabilised the keto-enol form. Electron-donating groups generally stabilised the keto-enol form. Contrary to common expectations, a direct correlation between dipole moment and stability order was not universally observed within 1,2-cyclodione systems, with the NH2 group demonstrating nuanced effects. Similarly, dipole moment and electrostatic potential variance did not exhibit consistent correlation under the influence of different substituents. The diketo tautomer was generally found to be more reactive than the keto-enol form, especially in non-polar solvents. Surprisingly, substituent groups decreased the reactivity of the diketo form relative to the unsubstituted compound.

Introduction
The substituent effect is a crucial driver in the analysis of structure-reactivity relationships (SRRs). Several factors, including inductive, resonance (mesomeric), and polarisation effects (through-bond, through-space, and field effects), contribute to the impact of substituents on chemical reactivity. 1 Both the ‘through-bond’ and ‘through-space’ inductive effects exhibit a rapid decrease as the distance from the substituent increases. 2 The theory of substituent effects has its root in Robinson and Ingold’s foundational research. It primarily serves as a qualitative framework for elucidating and predicting experimental reactivity and stability. 3 The fundamental question can be articulated as follows: ‘How do two components of a molecule exert influence upon each other?’. Investigating tautomerism in organic compounds is a central focus in theoretical studies, utilising diverse quantum-chemical approaches. 4 Understanding the relative stability, particularly in the context of three- to seven-member 1,2-cyclodiones, and the subsequent conversions between tautomeric forms is crucial for both biologists and chemists. 5 While extensive experimental and theoretical research has explored tautomerisation as a fundamental process in bioorganic chemistry with implications for condensation reactions, 6 significant questions remain regarding the specific interactions between substituents and the 1,2-cyclodione ring system.
Cyclodiones, a class of organic compounds containing two ketone functional groups within a cyclic structure, have captivated chemists due to their unique reactivity and potential applications in organic synthesis. 7 These fascinating molecules exhibit a distinct propensity for keto-enol tautomerism, a phenomenon that plays a pivotal role in their reactivity and synthetic utility. Studies have shown that the enols produced from unhindered, unconjugated ketones are present in minimal amounts at equilibrium. 5 Gaining insight into the relative stabilities of tautomeric forms holds significance from a structural chemistry perspective. 8 Moreover, understanding the variations in tautomerisation energies across different derivatives provides valuable insight into the impact of substituents on molecular reactivity and stability.
Substituent groups are generally characterised by electron-realising and electron-withdrawing and are classified in terms of their inductive effects as +I and −I, respectively, and +M and −M, respectively, for the case of their resonance effects. 2 For example, based on our study, substituent groups, NH2 +I (inductive effect), CH3 +I (inductive effect), CF3 −I (inductive effect) and Cl −I (inductive effect). Our study substituent groups were selected in form strong and its corresponding weak in term of inductive effect.
Keto-enol tautomerisation (Figure 1) of 1,2-hexanedione (cyclohexane-1,2-dione)
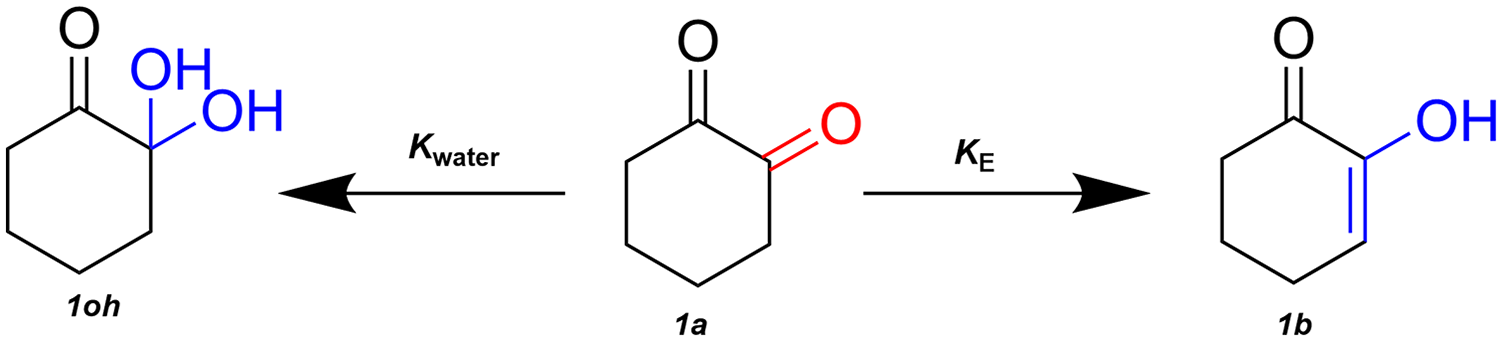
Two-dimensional molecular structures tautomerism in cyclohexane-1,2-dione.
In this article, we investigated the influence of substituents on the 1,2-cyclodiones system, comprising rings ranging from three to seven members. Specifically, we computationally examined derivatives of 1,2-cyclopropanedione (3a and 3b), 1,2-cyclobutanedione (4a and 4b), 1,2-cyclopentanedione (5a and 5b), 1,2-cyclohexanedione (6a and 6b), and 1,2-cycloheptanedione (7a and 7b) (refer to Figure 2). Our analysis focuses on exploring their molecular reactivity and stability at the same level of theory, considering both gas and solution phases. We accounted for solvation effects using water (ε = 78), acetone (ε = 21), and cyclohexane (ε = 2) as solvents. 11 The substituent groups under study include NH2, CH3, CF3, and Cl. These substituents were strategically selected for this study not only based on their inductive effects but also due their diverse electronic properties and relevance in the context of 1,2-cyclodiones. (1) Range of inductive effects is on the chosen substituents encompass a range of inductive effects, from the strongly electron-donating NH2 group to the strongly electron-withdrawing CF3 group. This allows for a systematic investigation of how the inductive effect influences keto-enol tautomerism and reactivity. (2) Resonance effects occurs to some of these substituents also exhibit significant resonance effects. For instance, the NH2 group can donate electron density through resonance, while the Cl group can withdraw electron density through resonance. Including substituents with varying resonance capabilities allows for a more comprehensive exploration of electronic effects on tautomeric equilibria and reactivity, (3) Prevalence in related compounds to the selected substituents are commonly found in various organic compounds, including natural products and pharmaceuticals containing 1,2-diketone motifs. Therefore, understanding their influence on the tautomeric behaviour and reactivity of 1,2-cyclodiones has broader implications for the design and synthesis of novel compounds with tailored properties. (4) Mechanistic insights to the diverse electronic properties of the chosen substituents enables a deeper exploration of the mechanistic factors governing keto-enol tautomerism. For example, the unique ability of the NH2 group to stabilise the keto-enol form, as observed in this study, prompts further investigation into potential resonance or hydrogen bonding interactions that might contribute to this stabilisation. In summary, the selection of NH2, CH3, CF3, and Cl as substituents was motivated by their diverse electronic properties, prevalence in related compounds, and potential to provide mechanistic insights into the keto-enol tautomerism of 1,2-cyclodiones. This comprehensive approach allows for a more thorough and impactful investigation of substituent effects on the reactivity and stability of these intriguing molecules.

The optimised geometrical structures in keto-enol tautomerisation process of substituted cyclic-1,2-diones of the three- to seven-membered rings.
Computational methods
All calculations were conducted using the Maestro Materials Science Suite. 12 For accuracy, the three-dimensional (3D) molecular geometries of the 1,2-cyclodiones system (Figure 2) were initially generated with the Maestro editor in the Schrödinger Materials Science Suite. We then searched for the lowest energy conformer for each compound using the OPLS4 force field on the MacroModel module. 13 This step ensured the desired molecular symmetry and proximity to a minimum. 14
The identified lowest energy conformers underwent further optimisation in the gas phase through DFT methods. Jaguar 15 programme, as implemented in Schrödinger Materials Science Suite, was employed for DFT calculations, specifically for conducting geometry optimisations and vibrational frequency analyses. The calculations were carried out using the M06-2X/6-311G++** level of theory, focusing on the gas phase. The frontier molecular orbital properties, atomic electrostatic potential (ESP) charges, and dipole moments were also calculated at the same theoretical level.
DFT is based on the Hohenberg and Kohn 16 theorem, which states that the ground-state energy of a many-electron system can be uniquely expressed as a functional of the electron density, allowing for efficient computational calculations of electronic properties. DFT is a vital method for predicting molecules’ chemical reactivity and thermodynamic properties. 17 It has proven highly successful in establishing the theoretical foundations of well-known qualitative chemical concepts.
To assess the impact of the solvent environment, the geometries of the tautomers were re-optimised in three different solvents, that is, water, acetone, and cyclohexane, maintaining the same theoretical level. To assess solvent effects on the relative stability and geometry of the 1,2-cyclodiones system, we employed the self-consistent reaction field (SCRF) method. For this, we used the polarisable continuum model (PCM) with the conductor-like polarisable continuum model (C-PCM) variant. Studies have shown that the C-PCM variant is a reliable and efficient method for computing solvation energies, structures, and properties of molecules in solution. 18
Results and discussion
Energy and thermodynamic parameters
The obtained energies difference, enthalpy change and Gibbs free energies difference for the two main possible 1,2-cyclodiones system (tautomer) are listed (see Table 1) calculated using M06-2X/6-311G++** and CPCM/ M06-2X/6-311G++** methods.
Calculated thermodynamic properties simulated with M06-2X/6-311G++** for a vacuum (gas) and different solvent (water, acetone, and cyclohexane) ΔH,

In the 1,2-cyclopropanedione system (3a and 3b), the enol form, 2-hydroxy-2-cyclopropen-1-one, is of considerable chemical interest. This interest stems from its role as a photochemical precursor to hydroxyacetylene, formed through the photodecarbonylation of hydroxycyclopropenone, this study confirms that the keto-enol form exhibits greater stability than the diketo form due to its enhanced aromatic character.5,10 Calculated Gibbs free energies (Table 1) further demonstrate that substituent groups on the 1,2-cyclopropanedione system maintain the stability of the keto-enol form in both gas and solution environments. These energies (in kcal/mol) range from −12.0 to −15.9 (H), −25.0 to −29.4 (NH2), −15.7 to −20.1 (CH3), −10.4 to −14.7 (CF3), and −13.6 to −17.9 (Cl) both in the gas phase and solution. The observed stability order (NH2 > CH3 > H > Cl > CF3) highlights the stabilising effect of electron-donating substituents. Notably, the keto-enol form exhibits its highest stability in the water phase compared to other phases (i.e. gas, acetone, and cyclohexane phase).
The 1,2-cyclobutanedione system (4a and 4b), particularly the diketo form, 1,2-cyclobutanedione, holds particular interest in thermal decomposition studies because it adopts a locked cis configuration. 19 The scientific literature confirms that the diketo form is more stable than the keto-enol form in this system. This stability difference is attributed to the increased angle strain in the keto-enol form. With three sp 2 -hybridised carbon atoms, the four-membered ring experiences more strain compared to the diketo form, which has only two sp 2 -hybridised carbons. 5 Calculated Gibbs free energies (Table 1) show that diketo form derivatives (4a) are more stable than their keto-enol counterparts. Stability differences range from 6.07 to 6.46 kcal/mol (H), 2.70 to 2.86 kcal/mol (CH3), 8.22 to 8.35 kcal/mol (CF3), and 2.03 to 2.62 kcal/mol (Cl), both in gas phase and solution. Interestingly, the presence of an NH2 substituent stabilised the keto-enol form by −10.07 to −10.97 kcal/mol in both gas and solution phases. These results indicate that electron-donating substituents increase the stability of the keto-enol form. The observed stability order (CF3 > H > CH3 > Cl > NH2) highlights that electron-withdrawing substituents play a major role in substantially enhancing the preference for the diketo form (4a) over its keto-enol counterpart.
In contrast to smaller ring systems, the cyclopentane-1,2-dione system (5a and 5b), the keto-enol form derivatives (5b) demonstrate greater stability than their corresponding diketo forms. Stability differences (in kcal/mol) range from −0.48 to 0.68 (H), −15.91 to −16.14 (NH2), −1.35 to −3.16 (CH3), −0.96 to −3.26 (CF3), and −5.21 to −8.14 (Cl), measured in both the gas phase and in solution. The observed stability order of the keto-enol form is as follows: NH2 > Cl > CH3 > CF3 > H. Interestingly, the unsubstituted cyclopentane-1,2-dione from Table 1 suggests that the keto-enol form is favoured in polar solvents like water and acetone, while the diketo form gains prominence in non-polar solvents (cyclohexane) and the gas phase. This highlights the significant role solvent polarity plays in determining the stability of the unsubstituted five-ring system.
In the cyclohexane-1,2-dione system (6a and 6b), the keto-enol form (6b) demonstrates a thermodynamic preference over the diketo form (6a). Literature reports attribute this to unfavourable lone pair repulsion in the diketo form, leading to higher energy. 9 This preference is observed even for the unsubstituted compound. Substituted keto-enol form derivatives (6b) further demonstrate greater stability than their diketo counterparts. Stability differences (in kcal/mol) range from −0.86 to −4.01 (H), −14.85 to −16.04 (NH2), −4.09 to −6.81 (CH3), −0.013 to −3.67 (CF3), and −3.49 to −7.31 (Cl), measured in both the gas phase and in solution. The observed stability order of the keto-enol form is as follows: NH2 > Cl > CH3 > H > CF3. Interestingly, the keto-enol form exhibits its highest stability in the gas phase compared to solvents, regardless of polarity (e.g. water, acetone, and cyclohexane). This gas-phase preference could be due to the lack of solvent interactions that might destabilise the keto-enol form. In cycloheptane-1,2-dione system (7a and 7b), theoretical reports suggest that the unsubstituted diketo form (7a) exists in a puckered conformation. This conformation offers greater stability compared to the corresponding keto-enol form (7b), which adopts an envelope-like structure and experiences increased ring strain. 5 The stability of the diketo form is further enhanced by its two carbonyl groups being oriented opposite to each other, minimising lone pair repulsion. This stability trend holds true for most substituents, favouring the diketo form as visualised in Figure 3. Interestingly, the NH2 substituent is an exception to this trend. Here, the keto-enol form is thermodynamically favoured, being approximately 8 kcal/mol more stable than the diketo form.
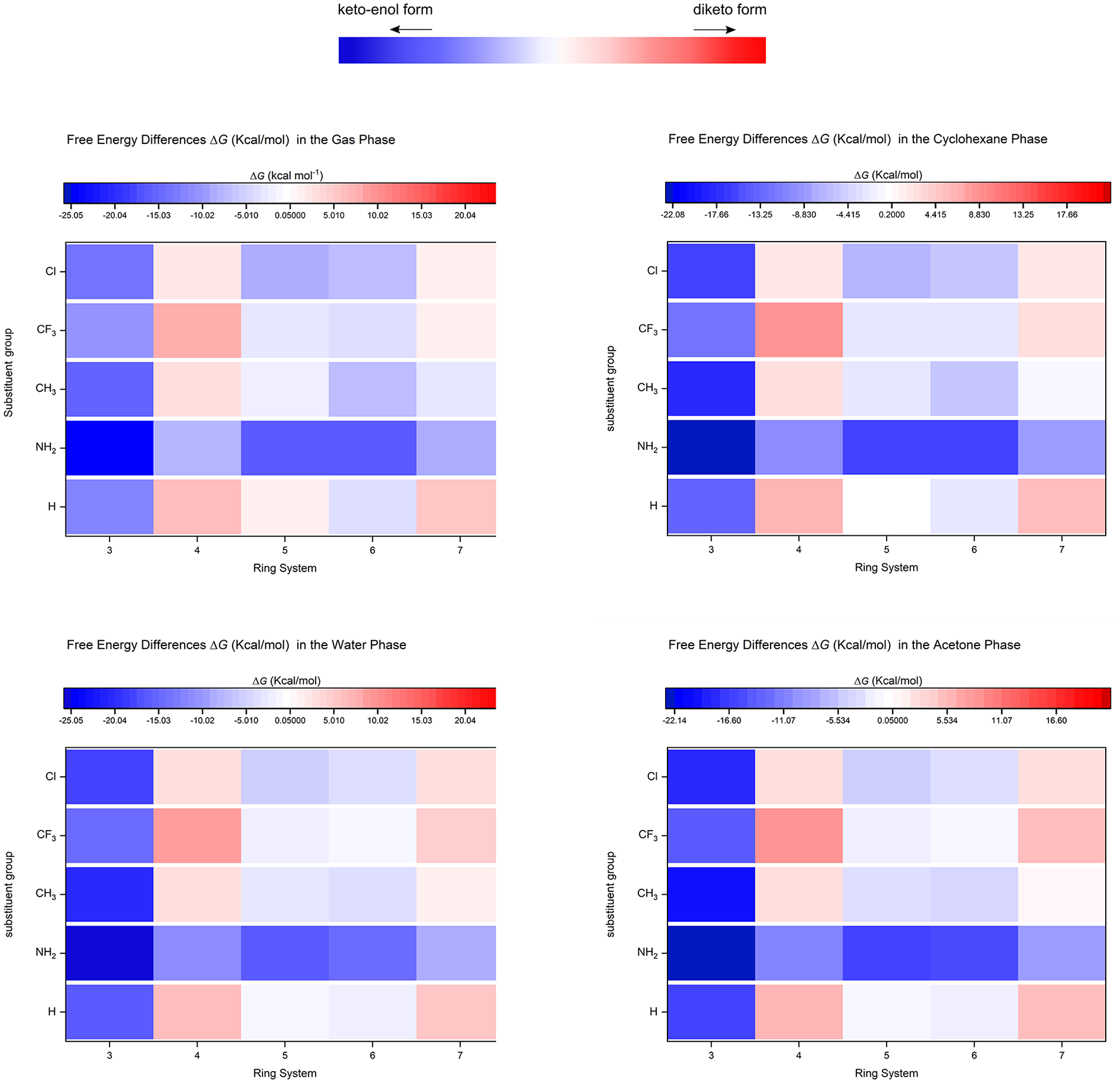
Change in Gibbs free energy in (kcal/mol) of the 1,2-cyclodiones system (tautomer) in vacuum (gas) and different solvent (water, acetone, and cyclohexane).
The studied compounds exhibit ring-size-dependent tautomerism. Three-, five-, and six-membered ring systems favour the keto-enol form, whereas four- and seven-membered rings tend to exist in the diketo form. Notably, an NH2 substituent stabilises the keto-enol tautomer exclusively. Figure 3 visually demonstrates how electron-withdrawing groups (EWGs) destabilise the keto-enol form. This destabilisation arises from the EWG weakening the crucial C=O double bond in the keto form and disrupting the favourable electron distribution in the enol form. In contrast, electron-donating groups (EDGs) stabilise the keto-enol form by strengthening the enol’s ability to accommodate a negative charge.
Influence of amino group to the keto-enol form
Depending on the molecule structure and environment, the −NH2 (amino) substituent can stabilise the keto-enol tautomeric equilibrium through many methods. (a) The hydrogen bonding mechanism can uniquely stabilise the keto-enol form, whereby the NH2 group can make hydrogen bonds with either the oxygen atom of the carbonyl group (in the keto form) or the hydroxyl group in the enol form. Within the enol form, the NH2 group can establish intramolecular hydrogen bonds with the hydroxyl (−OH) group, thereby stabilising the enol form (b) electron donation: an NH2 group donates electrons by resonance or inductive actions. In the enol form, a C=C bond and a −OH group allow the NH2 group to contribute electron density to the system. By increasing the electron density on the carbon–carbon double bond, the stability of the enol form can be enhanced. (c) Resonance stabilisation: Under certain circumstances, the NH2 group can resonate with the conjugated π-system of the enol form. The electron density delocalisation phenomenon between the NH2 group and the enol conjugated system can enhance the enol form’s stability.
For instance, the compound 3-amino-2-hydroxycyclohept-2-en-1-one (Figure 4) consists of a cycloheptenone (a seven-membered ring with a keto group) that has both an amino (−NH2) group and a hydroxyl (−OH) group. Because of both electrical and steric considerations, this particular structure enables the NH2 group to stabilise the enol form uniquely. An NH2 group might stabilise the enol form in this system as follows: the keto ester has a carbonyl group (C=O) at position 1, an amino group (−NH2) at position 3, and a hydroxyl group (−OH) at position 2. The formation of the enol form occurs when a proton is transferred from the alpha-carbon to the carbonyl oxygen, forming a hydroxyl (−OH) group at position 1 and a C=C double bond between carbons 1 and 2. The stabilisation processes involving NH2 in the enol form occur through the following pathways (1) intramolecular hydrogen bonding occurs when the −OH group at position 2 in the enol form forms a hydrogen bond with the NH2 group. This hydrogen bond stabilises the enol form by decreasing the system’s total energy and (2) resonance stabilisation involves the donation of electron density by the NH2 group through resonance. The lone pair on nitrogen in the enol formulation can resonate with the neighbouring C=C bond and the −OH group. This prolonged conjugation enhances the stability of the enol form by distributing electron density over the ring. (3) Electron donation and inductive effects: The NH2 group functions as an EDG by means of both resonance and inductive influence. It raises the electron density on the neighbouring carbon; therefore, enhancing the electron-richness and stability of the C=C bond in the enol form as against the keto form, (4) Steric effects occur in the keto form when there is steric repulsion present between the NH2 and the carbonyl group, which is caused by their close closeness. Reduction of this repulsion in the enol form raises the favorability of the enol form.

Structure of 3-amino-2-hydroxycyclohept-2-en-1-one.
Based on the above example, the presence of the NH2 group at position 3 enhances the stabilisation of the enol form compared to the keto form. Within this molecule, the stability of the enol tautomer is enhanced by the interaction of intramolecular hydrogen bonding and resonance between the amino and enol groups. Fundamentally, including the amino group at the third position in 3-amino-2-hydroxycyclohept-2-en-1-one promotes the stabilisation of the enol form mainly through intramolecular hydrogen bonding and resonance stabilisation.
Atomic ESP charges and dipole moments
Atomic ESP and dipole moment (μ) are fundamental physicochemical properties that offer valuable insights into molecular structure and reactivity.20,21 Understanding dipole moments (μ) is crucial in the study of tautomerism because distinct tautomeric forms often exhibit differences in their electronic structures, leading to potentially significant variations in dipole moments. 22 The ability to predict dipole moments facilitates the assessment of tautomer stability in polar environments. 23 ESP maps visualise the distribution of charge within a molecule, pinpointing regions of positive and negative potential that correspond to electrophilic and nucleophilic sites, respectively, and thereby influencing reactivity.
The

where

To calculate the total charge density of a molecule, we consider the contributions from each atom:

where
The ESP of a molecular system is directly determined by its total charge density distribution. It can be calculated by integrating the contributions from all infinitesimal charge elements:

where
To assess the overall variability of ESP across the molecular surface, we calculate the ESP total variance

where
A high ESP total variance
The dipole moment (μ) provides insights into the overall charge separation within a molecule and how it might orient itself in a solvent environment. It is calculated as:

where
Theoretical calculations at the M06-2X/6-311G++** level of theory was employed to analyse both ESP and dipole moments in the study of keto-enol tautomerism within 1,2-cyclodiones of varying ring sizes. These analyses shed light on how substituents affect, the stability, and reactivity of different tautomeric forms. By examining ESP variance (ESP total variance
The calculated ESP variance and dipole moment data (see Table 2) reveal differences between keto-enol and diketo forms. Both ESP variance and dipole moment increase when transitioning from gas to solution, with the highest dipole moments observed in water, the most polar solvent. This trend holds true for both tautomers.
Calculated dipole moment (D), ESP variance in (kcal/mol)²), energies of the HOMO and LUMO orbitals in (a.u) and HOMO/LUMO energy band gaps (in eV).

Small rings (three- and four-membered): For three-membered rings in the gas phase, the keto-enol form exhibits a higher value of dipole moment compared to the diketo form, indicating that greater polarity is associated with a wider range of ESPs. Notably, the Cl-substituted keto-enol form displays a high dipole moment (5.0113 D) and a significantly elevated ESP variance (473.9535 (kcal/mol))² while the NH2-substituted keto-enol form displays a high ESP variance (511.8385 (kcal/mol))² and a dipole moment (4.3153 D). Furthermore, the observed dipole moment order of the keto-enol form is Cl > NH2 > CF3 > CH3 > H, while the ESP variance order is NH2 > Cl > CF3 > H > CH3 (in both gas and solution phases). This suggests a lack of direct correlation between dipole moment and ESP variance order under the influence of different substituent groups. In the four-membered rings (gas phase), the diketo form tends to exhibit a higher dipole moment than its keto-enol counterpart. This trend runs counter to expectations since the keto-enol form typically displays higher ESP variance. EWGs like CF3 and Cl in the diketo form display high ESP variances [174.9059 and 174.0080 (kcal/mol)², respectively]. Conversely, EDGs such as NH2 and CH3 exhibit high dipole moments (4.5011 D and 4.2574 D, respectively). The observed dipole moment trend order in the keto-enol form is NH2 > CH3 > H > Cl > CF3, while the ESP variance order is CF3 > Cl > NH2 > H > CH3. This pattern holds true in both gas and solution phases. This study suggests a direct correlation between dipole moment and stability order in these tautomers, a relationship that runs counter to the trend observed with ESP variance.
Intermediate rings (five- and six-membered), in five-membered rings, a distinct pattern emerges: EDGs induce a high dipole moment in the keto-enol form, whereas EWGs lead to a high dipole moment in the diketo form. In addition, the keto-enol form typically displays higher ESP variance. In six-membered rings, results demonstrate a greater dipole moment in the diketo form compared to its keto-enol counterpart. This observation extends to ESP variance, which also appears dominant in the diketo form. Notably, EDGs induce a high dipole moment, specifically within the diketo form. This study highlights a lack of direct correlation between dipole moment and order of stability in these tautomers. Surprisingly, the least stable tautomer (diketo form) exhibits the largest dipole moment in both gaseous and aqueous phases, a pattern is also mirrored in ESP variance. The findings are supported by the study conducted by Anandan et al., 25 indicates that there is no direct correlation between dipole moment and order of stability under SCRF conditions among the studied tautomers.
In seven-membered rings, a direct correlation between dipole moment and order of stability generally exists within these tautomers. However, the NH2 group stands out as a potent outlier. It exhibits a greater dipole moment in the keto-enol form (7.5946 D) within a water phase compared to its diketo counterpart. This pattern remains consistent in acetone but differs in cyclohexane and gas phases. Interestingly, unsubstituted seven-membered rings display the highest ESP variance, followed by those with EDGs. These findings reveal a complex interplay between ring size, substituent effects, and solvent polarity in determining dipole moments and ESP variances of diketo and keto-enol tautomers. Interestingly, a direct correlation between dipole moment (see Figure 5) and order of stability is not universally observed. This is particularly evident in the presence of potent outliers such as the NH2 group, further highlighting the nuanced behaviour of these tautomeric systems.

Dipole moments in (Debye) of the 1,2-cyclodiones system (tautomer) in vacuum (gas) and different solvent (water, acetone, and cyclohexane).
ESP maps were generated (see Figure 6) to visualise electron density distribution within molecular structures, elucidating their reactivity. In the gas phase, the NH2-substituted diketo form within a four-membered ring presents an intriguing contradiction: it exhibits a high dipole moment but a surprisingly low ESP variance. This discordance highlights a potential disharmony between these properties. Comparisons between keto-enol forms in a three-membered ring, using NH2 and Cl substituents, emphasise substituents’ profound impact on both dipole moments and ESP variances. Further investigations reveal that the Cl-substituted keto-enol form displays marked shifts in these properties across different solvent phases, underscoring the critical role of solvent environments in molecular behaviour. Similarly, explorations of the NH2-substituted keto-enol form within a seven-membered ring showcase notable variations in dipole moment across various solvents, providing further insights into solvent effects. These analyses of ESP distributions yield valuable knowledge regarding the structural characteristics and reactivity of the molecules studied, advancing our chemical understanding.

Electrostatic potential (ESP) maps of the optimised geometrical molecular structures.
Correlation matrix: HOMO-LUMO gap and ESP values
Theoretical calculations at the M06-2X/6-311G++** level of theory was employed to analyse HOMO and LUMO energies in the study of keto-enol tautomerism within 1,2-cyclodiones of varying ring sizes. These calculations provide insights into the reactivity and stability of both diketo and keto-enol tautomers across different phases. The data suggest that the diketo tautomer generally exhibits higher reactivity than the keto-enol form, primarily due to its increased electrophilicity, which arises from the presence of two conjugated carbonyl groups. For example, in the gas phase, the diketo form of cyclopropane-1,2-dione demonstrates a smaller HOMO-LUMO energy gap compared to the keto-enol form, indicating greater reactivity. This pattern persists across other phases, such as water, acetone, and cyclohexane, though the exact influence of the solvent environment varies.
In the gas phase, there is a strong negative correlation between the HOMO-LUMO gap of the diketo form and its ESPmax (r = −0.5869), as well as with ESPmin (r = −0.6948) (Figure 7). These relationships suggest that as the reactivity of the diketo tautomer increases (indicated by a smaller HOMO-LUMO gap), the ability of the molecule to accept or donate electron density also increases. Similarly, for the keto-enol form in the gas phase, the HOMO-LUMO gap correlates negatively with ESPmax (r = −0.8496) and more weakly with ESPmin (r = 0.2307), indicating that reactivity is still largely governed by electron distribution. However, the correlation with electron donation (ESPmin) is less pronounced.

The correlation matrix: HOMO-LUMO gap and ESP values in vacuum (gas) and different solvents (water, acetone, and cyclohexane).
In water, acetone, and cyclohexane phases, the relationship between the HOMO-LUMO gap and ESP values follows a similar trend but with some variations (see Supplementary Table S3 for phase-specific data). In the water phase, the correlation between the HOMO-LUMO gap and ESPmax for the diketo form is less negative (r = −0.4985) compared to the gas phase, reflecting the stabilising influence of polar solvents, which can reduce reactivity. The same trend is observed in the acetone and cyclohexane phases, where the correlations are slightly less negative, again underscoring the impact of solvent polarity on molecular reactivity. For the keto-enol form, the HOMO-LUMO gap’s relationship with ESP values becomes even less pronounced in polar solvents like water and acetone, suggesting that these environments particularly stabilise the keto-enol form, likely through hydrogen bonding, thereby reducing its reactivity.
Overall, the diketo tautomer exhibits higher reactivity compared to the keto-enol tautomer, particularly in non-polar solvents and in the absence of electron-donating substituents. This finding aligns with the smaller HOMO-LUMO gaps observed for the diketo forms across all phases, indicating a consistent trend of increased reactivity. The solvent environment plays a significant role in modulating these relationships, with polar solvents stabilising the keto-enol form and decreasing its reactivity.
Frontier molecular orbitals
The highest occupied molecular orbital (HOMO) energy level significantly influences a molecule’s behaviour in chemical reactions. Specifically, the HOMO energy determines the molecule’s capacity to donate electrons; the higher the HOMO energy, the greater the molecule’s ability to act as an electron donor. Conversely, the lowest unoccupied molecular orbital (LUMO) energy indicates the ability to accept electrons. 26 The HOMO energy is directly correlated with ionisation potential, while the LUMO energy reflects electron affinity. 27 A larger energy gap between the HOMO and LUMO suggests higher kinetic stability and lower chemical reactivity. 28 This is because adding electrons to a high-lying LUMO or removing them from a low-lying HOMO becomes energetically less favourable. Therefore, the magnitude of the HOMO/LUMO gap offers valuable insight into a molecule’s reactivity patterns.
Theoretical calculations at the M06-2X/6-311G++** level of theory was employed to analyse HOMO and LUMO energies in the study of keto-enol tautomerism within 1,2-cyclodiones of varying ring sizes. Results indicate (see Table 2) that the diketo tautomer exhibits higher reactivity than the keto-enol form due to its increased electrophilicity, stemming from the presence of two conjugated carbonyl groups. For example, in the gas phase, cyclopropane-1,2-dione’s diketo form demonstrates a smaller HOMO-LUMO energy gap (ΔE) compared to the keto-enol form (6.240 vs 8.112 eV). This smaller gap indicates greater reactivity. However, some studies indicate that the keto-enol form is more thermodynamically stable, likely due to a degree of aromaticity that can be gained in the enol form. 10 The solvent environment significantly influences reactivity; polar solvents stabilise the keto-enol form through hydrogen bonding, making it less reactive. This is seen in water where cyclopropane-1,2-dione’s keto-enol form has a wider energy gap than in the gas phase (8.782 vs 8.113 eV). While electron-donating substituents like alkyl groups increase electron density on the carbonyl carbon, they unexpectedly decrease the reactivity of the diketo form. This is evident in 3-methylcyclopropane-1,2-dione, which exhibits a slightly wider energy gap in the gas phase compared to the unsubstituted compound (6.276 vs 6.240 eV). Overall, the diketo tautomer demonstrates higher reactivity compared to the keto-enol tautomer, particularly in non-polar solvents and in the absence of substituents. This increased reactivity is attributed to the presence of two conjugated carbonyl groups in the diketo form, which enhances its electrophilicity. The diketo tautomer was generally found to be more reactive than the keto-enol form, especially in non-polar solvents. This increased reactivity is attributed to the presence of two conjugated carbonyl groups in the diketo form, which enhances its electrophilicity. The smaller HOMO-LUMO energy gap observed for diketo tautomers, particularly in non-polar solvents, further supports this conclusion. This work does not explicitly explore the potential applications or implications of this enhanced reactivity although the discussion could be expanded to include (1) synthetic applications where the increased electrophilicity of the diketo form in non-polar solvents could make it a useful substrate in various organic reactions, such as Diels-Alder cycloadditions or nucleophilic additions. Highlighting these potential synthetic applications would demonstrate the practical relevance of the findings (2) Biological implications is if 1,2-cyclodiones or related compounds are found in biological systems, the preferential reactivity of the diketo form in non-polar environments (e.g. lipid membranes or hydrophobic protein pockets) could have implications for their biological activity or metabolic transformations. Discussing such possibilities would broaden the impact of the research (3) further research directions to the observed reactivity trends could inspire future studies explore the use of diketo tautomers as reactive intermediates or catalysts in non-polar solvents. Suggesting these potential research directions would demonstrate the potential for further development based on the current findings.
Conclusion
This investigation into the effect of substituents on the 1,2-cyclodiones system revealed how substituent groups influence stability and reactivity. The studied compounds exhibit ring-size dependent tautomerism, with the NH2 substituent uniquely stabilising the keto-enol form. Surprisingly, dipole moment does not always directly correlate with stability, demonstrating the complex interplay of ring size, substituent effects, and solvent polarity. Findings suggest the diketo tautomer is generally more reactive than the keto-enol form, especially in non-polar solvents and without the influence of substituents.
Supplemental Material
sj-docx-1-chl-10.1177_17475198241305254 – Supplemental material for A density functional theory study of keto-enol tautomerism in 1,2-cyclodiones: Substituent effects on reactivity and thermodynamic stability
Supplemental material, sj-docx-1-chl-10.1177_17475198241305254 for A density functional theory study of keto-enol tautomerism in 1,2-cyclodiones: Substituent effects on reactivity and thermodynamic stability by Luqman Idd Juma and Rene Costa in Journal of Chemical Research
Footnotes
Acknowledgements
The authors are gratefully acknowledging the support from Dr Joseph Kyobe of the chemistry department, at St. John’s University of Tanzania for technical assistance during this study.
Author contributions
All authors have participated in (a) conception and design or analysis and interpretation of the data, (b) drafting the article or revising it critically for important intellectual content, and (c) approval of the final version of the manuscript document.
Availability of data and materials
This is upon the request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics declarations
The authors declare that no violation of ethics conscious committed or will be intended.
Consent to participate
Not applicable – This study involved computational chemistry methods only, with no human subjects.
Consent for publication
Not applicable – This study contains no personal or identifiable data.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.