
Abstract
2,4-Difluoronitrobenzene has been reacted with a linker diamine (ethylenediamine or propylenediamine) and butylamine, in either order, to give new molecular building blocks for porous supramolecular networks. Two structures were established by X-ray single crystal structure determinations. The propylenediamine structure displays small pores, which may be due to the longer and more flexible linker in the structure.
Introduction
The sequential reaction of 2,4-difluoronitrobenzene,

2,4-Difluoronitrobenzene,
Figure 2 shows that compound

Two examples of hydrogen-bonded hexamers. Left: compound
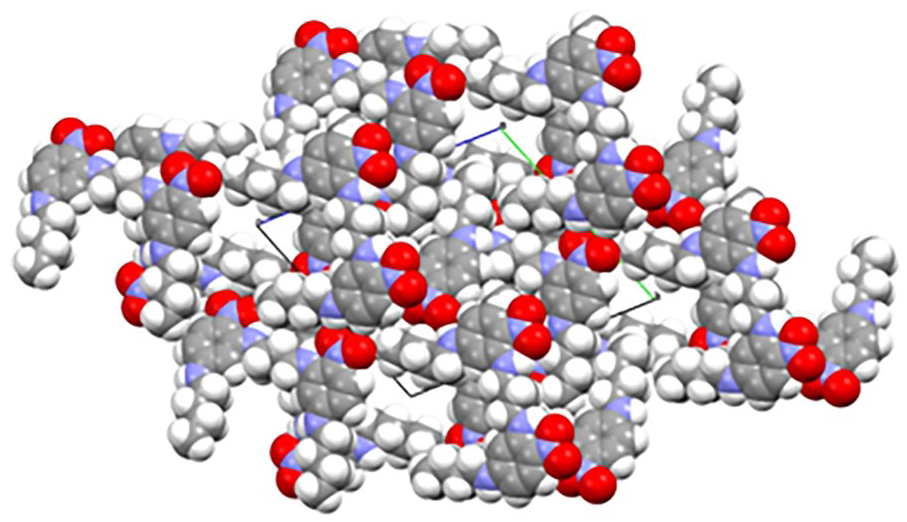
Left: hydrogen-bonded channels formed from compound
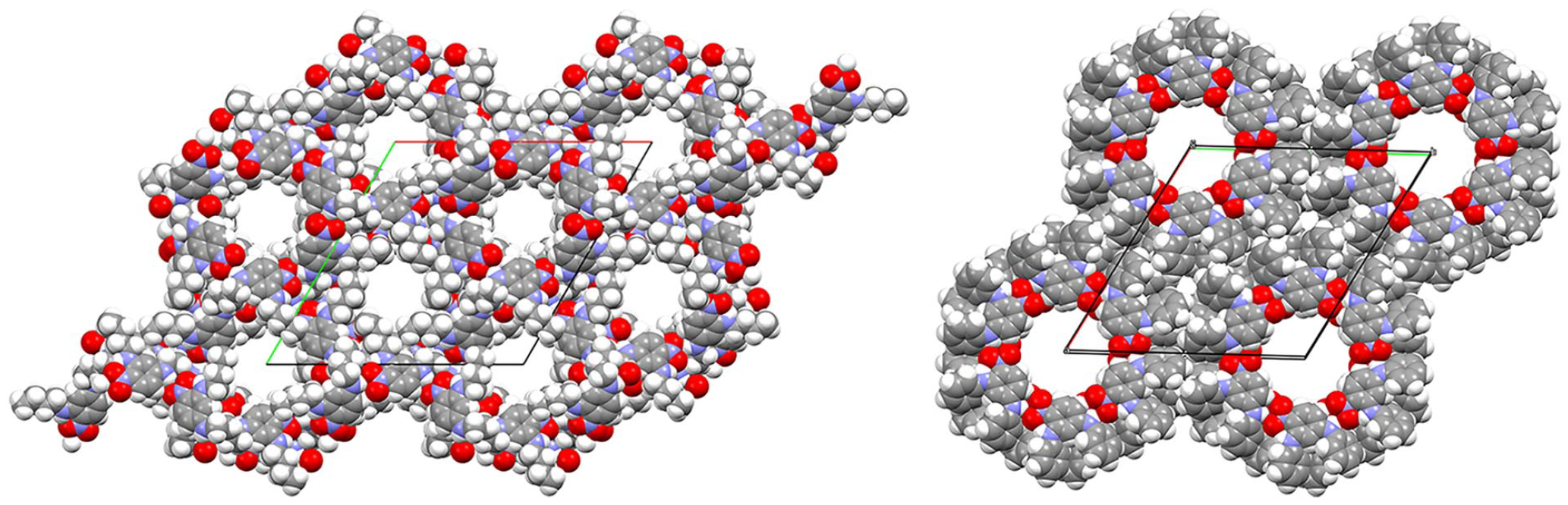
Figure 3 shows the packing diagrams from the X-ray single crystal structure determinations of compounds
These pores are empty apart from disordered solvent molecules. Unlike many clathrates, the existence of an open framework does not depend upon the inclusion of guest molecules. There are, however, a number of organic zeolites10–20 with potential for novel properties as well as metal-organic frameworks (MOFs)21–25 and materials which form 1D channels.26–35 These materials, known as nanoporous materials, with molecular-sized pores are of potential importance in molecular separation, heterogeneous catalysis, gas storage and carbon dioxide capture.
Discussion
Owing to the serendipitous discovery of porous structures formed from derivatives of 2,4-diaminonitrobenzene, such as compounds

One-pot synthesis of compound
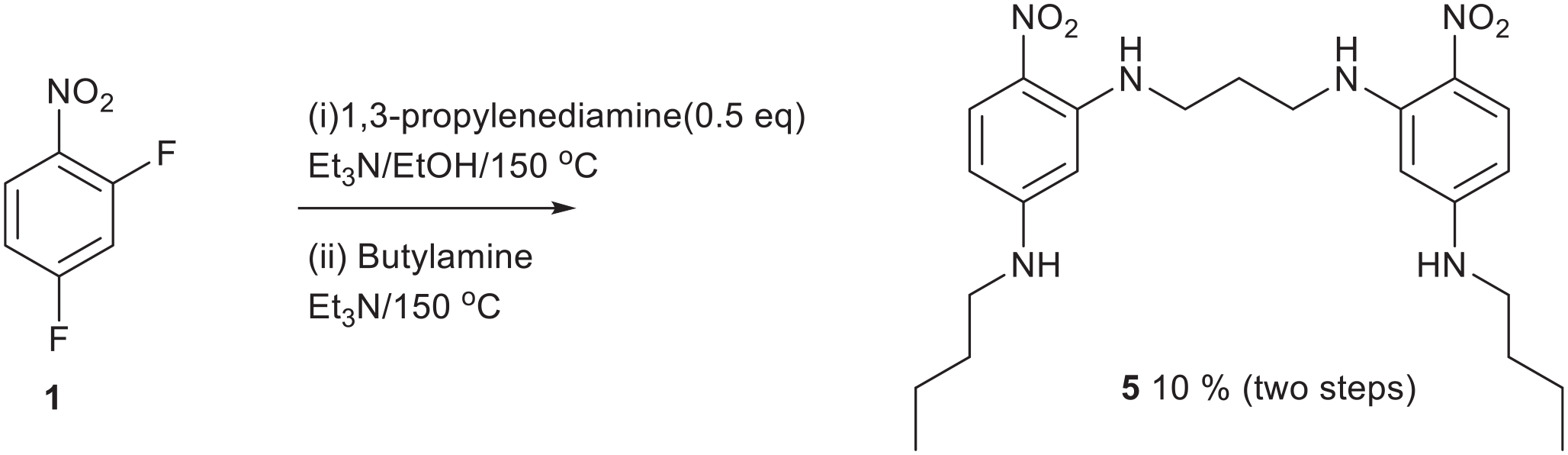
One-pot synthesis of compound

One-pot synthesis of compound
For reasons that are not clear, the ortho fluorine atom is more reactive to nucleophilic displacement than the para fluorine atom (Figure 4). The inductive and mesomeric activation of the ortho position must be stronger. Ethylenediamine first links two halves together followed by capping with butylamine at both ends. The first fluorine atom can be displaced under milder conditions but the second deactivated fluorine atom is best displaced in a pressure vessel at a higher temperature so both reactions were done together. These products are poorly soluble, which presented some problems with the acquisition of 13C NMR data in D6DMSO. Most peaks were observed but two weak, aromatic, signals were absent owing to poor solubility. The structure was confirmed with an X-ray single crystal structure determination (see below).
The synthesis of compound
Compound
The molecular structure of compound

The molecular structure of compound
The extended structure of compound
The molecular structure of compound

The molecular structure of compound
The extended structure of compound

Packing diagram of compound
The supplementary section has NMR charts for compounds 4, 5 and 6.
Conclusion
Further studies on the synthesis of diamino-substituted derivatives made from 2,4-difluoronitrobenzene
Experimental
IR spectra were recorded on a diamond-attenuated total reflection (ATR) Fourier transform infrared (FTIR) spectrometer. Ultraviolet (UV) spectra were recorded using a Perkin Elmer Lambda 25 UV-Vis spectrometer with EtOH as the solvent. The term sh means shoulder. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at 400 and 100.5 MHz, respectively, using a Bruker 400 spectrometer. Chemical shifts, δ, are given in ppm and measured by comparison with the residual solvent. Coupling constants, J, are given in Hz. High-resolution mass spectra were obtained at the University of Wales, Swansea, using an Atmospheric Solids Analysis Probe (ASAP) (positive mode) instrument: Xevo G2-S ASAP. Melting points were determined on a Cole-Palmer Stuart microscope.
Compound 4
2,4-Difluoronitrobenzene (250 mg, 1.57 mmol) in EtOH (10 ml) was mixed with ethylenediamine (47 mg, 0.79 mmol) and Et3N (159 mg, 1.57 mmol) in a Teflon-lined Parr pressure vessel. The vessel was heated at 150 °C for 12 h and then left to cool. An excess of BuNH2 (230 mg, 3.14 mmol) and EtOH (1 mL) were added. The vessel was heated for a further 12 h at 150 °C. After cooling, the contents of the vessel were diluted with DCM (100 mL) and extracted with water (150 mL) using a separating funnel. The DCM layer was collected, dried with MgSO4, filtered under gravity, then concentrated in vacuo. DCM, petroleum ether and EtOAc were investigated in various ratios and mixtures by TLC to find a good separation of the product from the other by-products. The product was purified by column chromatography on flash silica, using neat DCM to remove impurities then changing solvent to DCM/EtOAc (98:2) once the impurities were removed. Fractions 52–58 were collected. The crystals were then recrystallised in a large beaker by adding DCM and a small amount of petroleum ether and leaving the solvent to slowly evaporate. This produced the title compound (101 mg, 14 %) as orange blocks, mp 237–238 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 399 (log ε 4.1); νmax (diamond)(cm–1) 3335w, 1613s, 1546s, 1510s, 1469s, 1427s, 1409s, 1266s, 1220s, 1163s, 134s, 824s, 749s, 606s, 559s and 449s; δH (400 MHz; CDCl3) 0.91 (6H, t, J = 7.0), 1.36 (4H, s, J = 8.0), 1.53 (4H, q, J = 8.0), 3.08 (4H, q, J = 5.0), 3.62 (4H, s), 5.76 (2H, NH), 6.07 (2H, dd, J = 2.0 and 9.0), 7.09 (2H, t, J = 4.0 and 9.0), 7.81 (2H, d, J = 9.0 Hz) and 8.66 (2H, s, NH); δC (100.1 MHz; CDCl3) 14.2, 20.1, 30.9, 41.6, 42.2, 122.2, 128.6, 148.5 and 155.4) Two aromatic resonances are missing owing to poor solubility; m/z (Orbitrap ASAP) 445.2568 (M+ + H, 100%) C22H32N6O4H requires 445.2563.
Compound 5
2,4-Difluoronitrobenzene (250 mg, 1.57 mmol) in EtOH (10 mL) was mixed with 1,3- diaminopropane (58 mg, 0.79 mmol) and Et3N (159 mg, 1.57 mmol) in a Teflon-lined Parr pressure vessel. The vessel was heated at 150 °C for 12 h and then left to cool. An excess of BuNH2 (230 mg, 3.14 mmol) and EtOH (1 mL) were added. The vessel was heated for a further 12 h at 150 °C. After cooling, the contents of the vessel were diluted with DCM (100 mL), EtOAc (15 mL), and extracted with water (150 mL) using a separating funnel. The DCM layer was collected, dried with MgSO4, filtered under gravity and then concentrated in vacuo. The product was purified by column chromatography on flash silica using neat petroleum ether to remove impurities then changing solvent to petroleum ether/EtOAc (60:40) once the impurities were removed. Fractions 14–17 were collected and then recrystallised in a large beaker by adding DCM and a small amount of light petroleum ether. The solvent was left to slowly evaporate which yielded the title compound (73 mg, 10%) as yellow crystals, mp 149–150 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 401 (log ε 4.2); νmax (diamond)(cm–1) 3328w, 1614s, 1517s, 1544s, 1510s, 1456s, 1394s, 1312s, 1255s, 807s and 784s; δH (400 MHz; CDCl3)1.01 (6H, t, J = 8.0), 1.44 (4H, s, J = 8.0), 1.63 (4H, q, J = 8.0), 2.13 (2H, q, J = 6.0), 3.17 (4H, q, J = 6.0), 3.43 (4H, q, J = 6.0), 4.55 (2H, s, NH), 5.67 (2H, d, J = 2.0), 5.93 (2H, dd, J = 2.0 and 9.0), 8.03 (2H, d, J = 9.0) and 8.61 (2H, s, NH); δC (100.1 MHz; CDCl3) 13.8, 20.1, 28.4, 31.4, 39.9, 42.9, 90.0, 104.6, 123.6, 129.3, 148.1 and 154.7; m/z (Orbitrap ASAP) 459.2711 (M+ + H, 100%) C23H34N6O4H requires 459.2720.
Compound 6
2,4-Difluoronitrobenzene (250 mg, 1.57 mmol) in EtOH (10 mL) was added to a Teflon-lined Parr pressure vessel along with BuNH2 (115 mg, 1.57 mmol) and an excess of Et3N (318 mg, 3.14 mmol). The vessel was heated at 150 °C for 12 h and then left to cool. 1,3-Diaminopropane (58.2 mg, 0.79 mmol) and EtOH (1 mL) were then added. The vessel was heated at 150 °C for a further 12 h. Once cooled, the contents of the vessel were diluted with DCM (100 mL) and extracted with water (150 mL) using a separating funnel. The DCM layer was collected, dried with MgSO4, filtered under gravity, and then concentrated in vacuo. The product was purified by column chromatography on flash silica using DCM/EtOAc (95:5) and fractions 21–24 were collected. The crystals were then recrystallised in a large beaker by adding DCM and a small amount of light petroleum ether leaving the solvent to slowly evaporate. This gave the title compound (89 mg, 12 %) as orange-yellow crystals, mp 139– 140 °C (from dichloromethane:light petroleum ether). λmax (EtOH)/nm 404 (log ε 3.8); νmax (diamond)(cm–1) 3311s, 1616s, 1578s, 1540s, 1459s, 1396s, 1322s, 1252s, 1189s, 1160s, 808s, 751s, 652s and 544s; δH (400 MHz; D6DMSO) 0.91 (6H, t, J = 8.0), 1.36 (4H, s, J = 8.0), 1.59 (4H, q, J = 8.0), 1.87 (2H, q, J = 6.0), 3.19 (4H, q, J = 6.0), 3.25 (4H, q, J = 5.0), 5.67 (2H, s, NH), 6.08 (2H, dd, J = 2.0 and 9.0), 7.22 (2H, t, J = 4.0), 7.82 (2H, d, J = 9.0) and 8.47 (2H, NH); δC (100.1 MHz; D6DMSO) 14.0, 20.3, 27.8, 30.5, 42.0, 88.8, 106.5, 122.2, 128.8, 148.5 and 155.6 (one alkyl resonance is missing); m/z (Orbitrap ASAP) 459.2728 (M+ + H, 100%) C23H34N6O4H requires 459.2720.
Crystal-structure determinations
The crystal structures of
Crystal data for
Crystal data for
Supplemental Material
sj-docx-1-chl-10.1177_17475198241293518 – Supplemental material for Potential building blocks for porous organic materials
Supplemental material, sj-docx-1-chl-10.1177_17475198241293518 for Potential building blocks for porous organic materials by Michael John Plater, Maisie A Cowie and William TA Harrison in Journal of Chemical Research
Footnotes
Acknowledgements
We thank the UK EPSRC National Mass Spectrometry Service Centre for mass spectrometric data and the UK National Crystallography Centre (University of Southampton) for the X-ray data collections. Maisie Cowie performed all synthesis and obtained the characterisation data and WTA Harrison solved the crystallographic data sets. Data sets were obtained free of charge from the National Crystallography Center, Southampton University.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.