
Abstract
Four lignans, asarinin

Introduction
Zanthoxylum rhetsa (Roxb.) DC. belonging to the Rutaceae family, is a flowering and aromatic plant with a small wood, thorny bark of trunk and branches, and spicy fruits. This plant distributed in India, Thai Lan, Myanmar, Lao, and Vietnam.1,2 Z. rhetsa is an indispensable herb in Thai and Mong ethnic groups living in Northwest Vietnam, which use young leaves as a spice and fruits as the culinary soul of this region.1–3 In folk medicine, fruit, stem, or root barks are used to treat diarrhea, malaria, rheumatism, and worms; essential oil of fruits is used as an antibacterial agent and prevention of cholera. 1 Recent studies found that Z. rhetsa contains alkaloids, lignans, coumarins, triterpenoids,4–8 and monoterpenoids in the essential oil.9–12 In our previously publication, we have reported the chemical constituents and biological activities of essential oils from the barks, leaves, and fruits of this plant. 13 In this paper, we report the isolation and the structure elucidation, as well as the biological activities of secondary metabolites from the stem bark of Z. rhetsa.
Results and discussion
NMR analysis of secondary metabolites from stem bark of Z. rhetsa
The 1H NMR data of compounds

Recorded in aacetone-d6, b500 MHz; [18]: in CDCl3, d500 MHz.
13C NMR data of compounds

Recorded in aacetone-d6, c125 MHz; [18]: inCDCl3, e125 MHz.

Chemical structures of compounds
Compound
Cytotoxic activity of isolated compounds
Compounds
The results showed that compounds
The cytotoxic activity of compounds
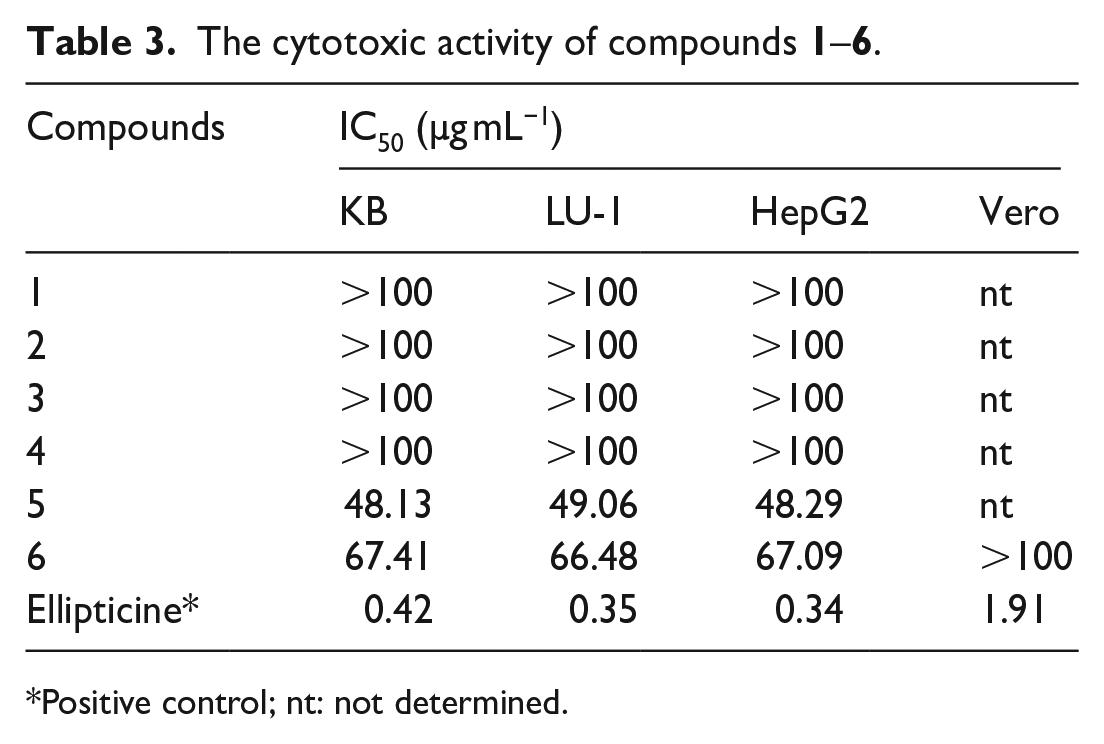
Positive control; nt: not determined.
Because hesperidin
Enzyme inhibitory activities of hesperidin (6)
Compound hesperidin
Enzyme inhibition of hesperidin

Positive control for the test of ACE-2 was MLN-4760; positive control for the test of Mpro was GC376; **in nM.
Docking using Autodock
Autodock4 is an open-source package that can predict rapidly the binding affinity of ligands toward a specific protein/enzyme target. In computation, binding free energy (ΔG) is calculated to estimate binding affinity of the ligand–target complex. It was reported by Gohlke et al. that ligand calculated partial charge with the PM6 method has been shown to greatly increase docking accuracy and cluster population of the most accurate docking. 23 According to the criteria of AutoDock4, lower binding free energy values indicate higher stability for the complex and favoring interaction of ligand within the binding site when it predicts the docking pose.24,25 Table 5 presents the docking results of studied compounds.
Docking results of hesperidin and reference ligand with lists of interacting residues.

As previous studies have reported, the active site of SARS-CoV-2 Mpro is constituted by key residues as follows: His41, Met49, Asn142, Gly143, Cys145, His164, Met165, Glu166, Lru167, Pro168, His172, Gln189, Thr190, and Ala191. 26 Binding conformation analysis of PF-07321332 indicate that five H-bonding were created by this compound with Cys145, Glu166, Arg188, Gln189, and Thr190, four among them are essential residues in the active binding region of SARS-CoV-2 Mpro. Besides, the interaction was further strengthened by hydrophobic bonds with His41, Met49, Pro52, Tyr54, His163, and Met165 (Figure 2). Regarding dock pose of hesperidin, the binding affinity of this ligand toward targeted protein was observed to be higher than the reference ligand (−14.36 and −12.41 kcal mol−1, respectively). The hydrophobic pockets formed with hesperidin involved residues His41, Met165, Pro168, and Ala191 while Leu141, Ser144, Cys145, Glu166, and Gln192 were the four essential residues creating hydrogen bonds with the ligand. These finding are in agreement with other studies published previously,27,28 thus, hesperidin could be assumed as a potential inhibitor of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Mpro.
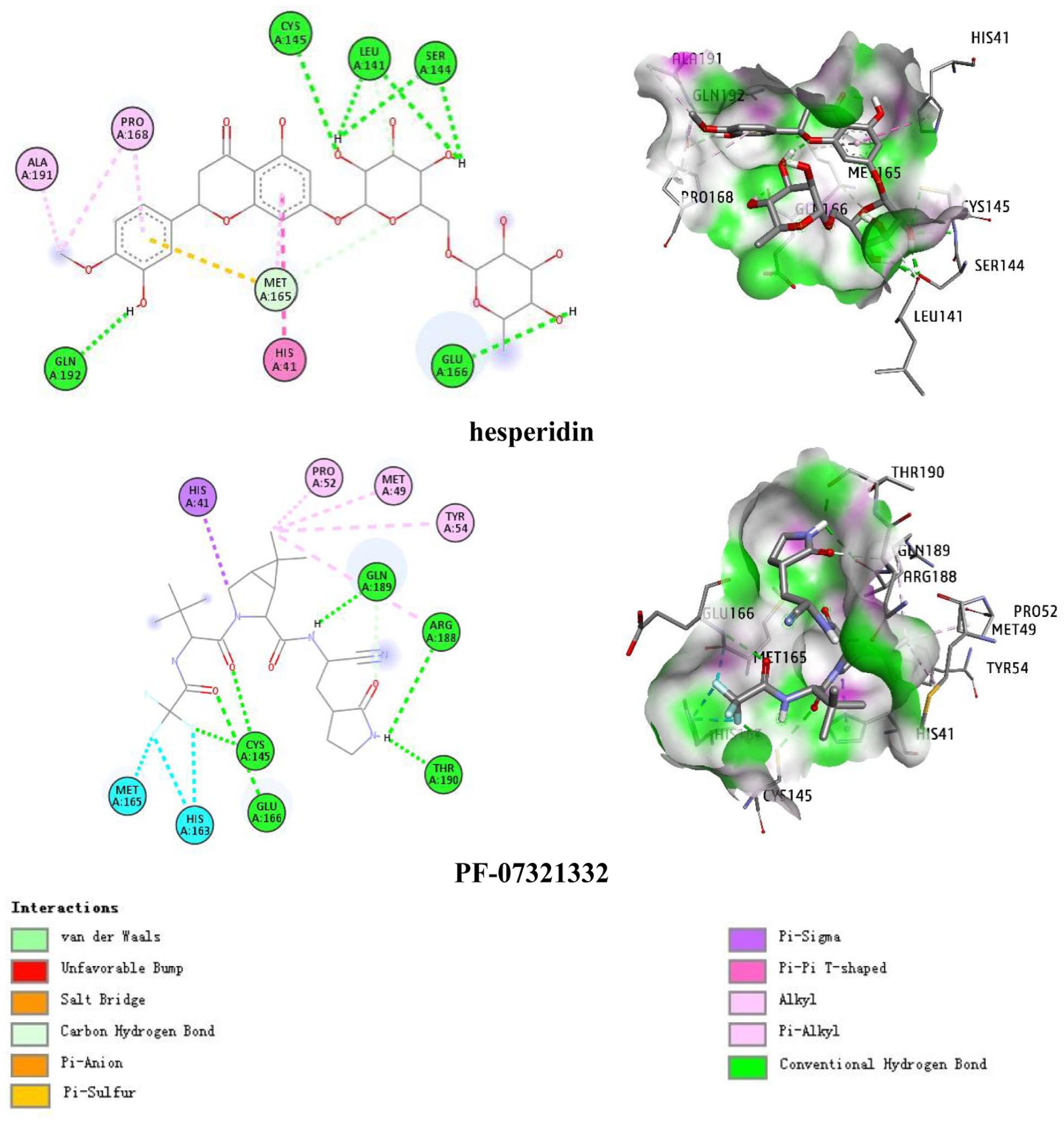
Docking conformation of hesperidin and PF-07321332 within the active site of SARS-CoV-2 main protease suggested by molecular docking simulation.
Conclusion
This study reports on the isolation, elucidation of chemical structures and cytotoxic activity of four lignans (asarinin
Experimental
General
The NMR spectra, including 1H NMR (500 MHz) and 13C NMR (125 MHz), DEPT, COZY (two-dimensional 1H–1H correlated spectroscopy), HSQC, and HMBC were recorded using a Bruker DRX 500 spectrometer. Column chromatography (CC) was performed on silica gel (Kieselgel 60, 70–230 mesh and 230–400 mesh, Merck) or RP-18 resins (30–50 μm, Fuji Silysia Chemical Ltd.). For thin-layer chromatography (TLC), pre-coated silica gel 60 F254 (0.25 mm, Merck) and RP-18 F254S (0.25 mm, Merck) plates were used.
Plant materials
The trunk bark of Z. rhetsa was collected at Thuan Chau, Son La, Vietnam, and identified by Dr Nguyen Quoc Binh, Vietnam Museum of Nature (VMN), Vietnam Academy of Science and Technology (VAST). The voucher specimen was deposited at the Institute of Natural Products Chemistry (INPC), VAST.
Extraction and isolation
The dried powdered stem bark of Z. rhetsa (5.0 kg) was extracted with hot methanol three times (each 10 L, 2 days) at 50 °C using an ultrasound-assisted technique, then removed solvent in vacuo to yield the methanol extract (270.0 g). In the methanol extract, there is a white solid. This solid was decanted and then purified on a reversed-phase RP-18 silica gel CC to yield compound
Asarinin
Horsfieldin
(5-(4-(3,4,5-trimethoxyphenyl)hexahydrofuro[3,4-c]furan-1-yl)benzo[d][1,3]dioxole)
5-(4-(3,5-dimethoxyphenyl)hexahydrofuro[3,4-c]furan-1-yl)benzo[d][1,3]dioxole
Piperonylic acid
Hesperidin
Syringin
β-sitosterol
Cytotoxic assays
The cell lines, including the KB (human oral carcinoma), HepG-2 (human hepatocellular carcinoma), and LU-1 (human lung carcinoma), were obtained from Milan University, Italy, and Hawaii University, USA. The human cancer cytotoxic activity of the compounds (
Enzyme inhibition assays
Compound

In which ΔRFU (S) and ΔRFU (EC) are the sample’s and control’s relative fluorescence units, respectively.
The inhibitory value is expressed as an IC50 value (the concentration of test agents that cause 50% inhibition), which was obtained by nonlinear regression using the TableCurve 2D program (SPSS Statistical Software, Chicago, IL).
Ligand preparation
The studied compounds were prepared three-dimensional structure using MarvinSketch version 19.27.0 and PyMOL version 1.3r1. 29 Energy minimization were then conducted using MM2 force field and quantum chemical calculation were performed using PM6 semi-empirical method implemented in Gaussian 09. 30 In this study, PF-07321332, a known inhibitor of SARS-CoV-2 Mpro, was selected as reference ligand. 31
Protein preparation
The X-ray crystal structure of SARS-CoV-2 Mpro was downloaded from the Protein Data Bank archive (PDB ID: 6LU7). 32 The protein structure was prepared using the Graphical User Interface program named Autodock Tools (ADT).
Docking using Autodock
The molecular docking study utilizes AutoDock 4.2.6 with Lamarckian genetic algorithm (LGA) for searching the optimum dock pose together with scoring function to calculate the binding affinity. ADT was employed to set up and performed docking calculation. In this study, we performed the docking study assuming that having a rigid protein and consider the conformational space of the ligands to analyze the inductive effect of the hybrid compounds. ADT was utilized to prepare protein for docking simulations. The heteroatoms including water molecules were deleted, and polar hydrogen atoms and Kollman charges were added to the receptor molecule. All other bonds were allowed to be rotatable. In the docking analysis, the binding site was enclosed in a box with the number of grid points in x × y × z directions and a grid spacing of 0.375 Å. Initially, AutoGrid was run to generate the grid map of various atoms of the ligands and receptor. After the completion of the grid map, AutoDock was run using autodock parameters as follows: GA population size, 300; maximum number of energy evaluations, 25,000,000; and the number of generations, 27,000. A maximum of 50 conformers were considered for each molecule, and the root mean square (RMS) cluster tolerance was set to 2.0 Å in each run. The outputs from AutoDock modeling studies were analyzed using PyMOL, 29 Discovery Studio Visualizer 33 , and LigPlus. 34 PyMOL was used to calculate the distances of hydrogen bonds as measured between the hydrogen and its assumed binding partner.
Supplemental Material
sj-docx-1-chl-10.1177_17475198231185704 – Supplemental material for Lignans and some other non-alkaloid compounds from the stem bark of Zanthoxylum rhetsa and their biological activities
Supplemental material, sj-docx-1-chl-10.1177_17475198231185704 for Lignans and some other non-alkaloid compounds from the stem bark of Zanthoxylum rhetsa and their biological activities by Tran Thi Tuyen, Pham Cao Bach, Do Huu Nghi, Pham Minh Quan, Pham Thi Hong Minh and Nguyen Thi Hong Van in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research is funded by Vietnam Academy of Science and Technology (VAST) under grant numbers NCVCC07.10/22-23 and ĐL0000.07/22-23.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.