Holliday’s industrial dye synthesis of 1865 was repeated by heating aniline hydrochloride with nitrobenzene and the products were characterised by modern spectroscopic methods. A red dye was isolated in low yield and characterised by an X-ray single-crystal structure determination as 9-phenyl-3-anilinophenazone-2-anil. The structure was identical to that made 93 years later in 1958 by the oxidation of 2-aminodiphenylamine hydrochloride with FeCl3 followed by heating the two products with aniline. An X-ray single-crystal structure determination was obtained for an isomer of anilinoaposafranine. A proposal is made that 9-phenyl-3-anilinophenazone-2-anil, isolated from Holliday’s synthesis, arises via the oxidation and dimerisation of 2-aminodiphenylamine with hot nitrobenzene so there is a common intermediate in both syntheses.
The molecular structure of John Holliday’s red dye is revealed by an X-ray single-crystal structure determination.
Introduction
o-Phenylenediamine 11–3 and 2-aminodiphenylamine hydrochloride 3 are easily oxidised in water by FeCl3.4–7 (Scheme 1) Kehrmann characterised the structure of compound 24 and reported the product 4 from the FeCl3 oxidation of compound 3.5,6 However, Fischer in contrast to this reported product 5 from the FeCl3 oxidation of compound 3.7,8 Degradation studies by Fischer confirmed that product 5 had the correct structure of this compound, which was known as anilinoaposafranine. Contributions were also made by Schopff and Ernst.9,10 In a later chapter of this fascinating molecular reactivity, compound 5 was prepared and its anti-tubercular activity was studied.11–14 This was an accidental discovery as solutions prepared from compound 3 showed increasing activity as slow oxidation to compound 5 occurred.11 It was shown that the products 4 and 5 are in fact both formed from the oxidation of compound 3 with either FeCl3 or p-benzoquinone but in differing ratios.14
The oxidation of some representative o-phenylenediamine derivatives with FeCl3.1–7
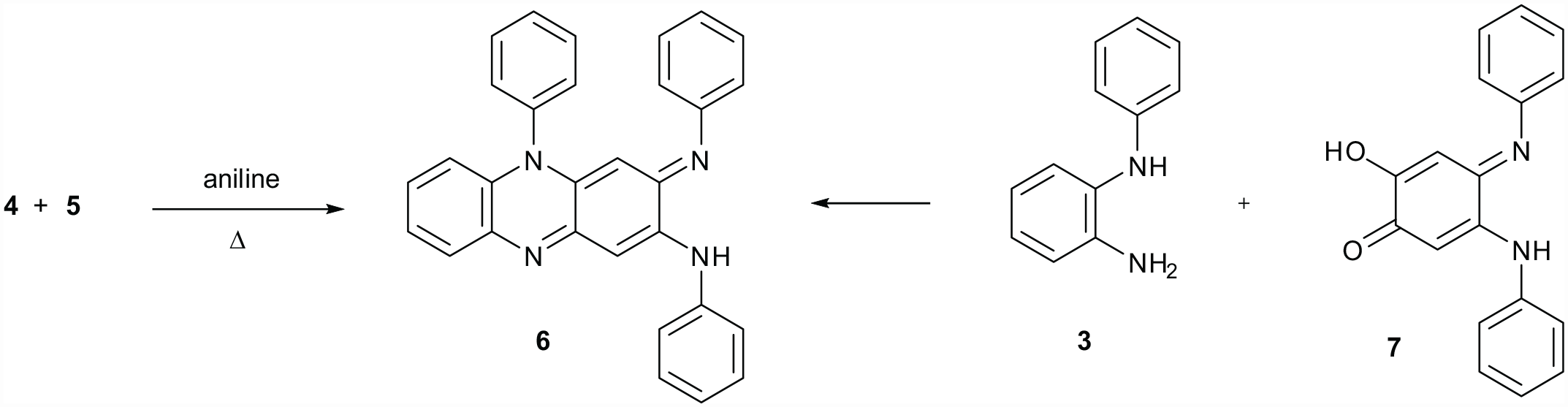
Twomey and co-workers showed that compounds 4 and 5 react in hot aniline to give compound 6.15 (Scheme 2). The same products were made by reacting compound 3 with diarylaminoquinone derivatives and these were explored for their biological activity16,17 (Scheme 2). The chemistry of compound 6 and its derivatives were extensively explored in the late 1950s by Twomey.18–22
The substitution of both compounds 4 and 5 with aniline to form compound 615 and an independent synthesis of compound 6.16,17
Discussion
Holliday’s industrial patent on dye synthesis,23 which forms the title of this publication, was uncovered by looking through the patent abridgement index24 and in Maurice Fox’s comprehensive book on Great Britain’s dye makers 1856–1976.25 Holliday reported a reaction, which involved heating 56 pounds of aniline hydrochloride and 44 pounds of nitrobenzene from which a red dye and a blue dye were isolated. No characterisation data were published, but the reaction was performed on a large scale. The reaction of nitrobenzene with aniline under basic conditions has been studied since this time but under these conditions, products were characterised.26,27 Holliday’s reaction was repeated by us, and we isolated a red and blue product albeit in very low yield (Scheme 3). The samples were pure and gave good mass spectral data. For the red compound, the relative molecular weight is 438.2 (C30H23N4) and for the blue compound, 453.2 (C30H24N5). Each structure has five benzene rings, and the long wavelength λmax (EtOH) absorbances were 360 and 574 nm, respectively. The blue compound was less soluble and poorly crystalline and was not studied further. It appears to have an extra primary amino group, which would explain the greater conjugation and blue colour. We obtained proton and carbon nuclear magnetic resonance (NMR) data for the red compound and carried out an X-ray single-crystal structure determination, which is shown in Figure 1.
A repeat of Holliday’s 1865 industrial dye synthesis.23
The molecular structure of compound 6 showing 50% displacement ellipsoids. The N–H···N hydrogen bond is indicated by a double-dashed line.
The asymmetric unit of compound 6 (Holliday’s method) contains one molecule (Figure 1). The central tricyclic ring system is slightly puckered as indicated by the dihedral angle between the C1–C6 and C7–C12 benzene rings of 5.74 (6)°; the central C1/C6/C7/C12/N1/N2 ring is almost planar (r.m.s deviation = 0.012 Å). The dihedral angles subtended by the pendant C13–C18, C19–C14 and C25–C30 phenyl groups with respect to the C1–C12/N2/N2 mean plane (r.m.s. deviation = 0.053 Å) are 35.58 (3), 68.39 (3) and 70.54 (3)°, respectively; the C19 and C25 rings are twisted in the same sense (dihedral angle = 12.18 (5)°), presumably to minimise steric repulsion. The mean bond length for C2–C3 and C5–C6 (1.362 Å) in the C1–C6 ring is much shorter than the mean of the other four bonds (1.455 Å), which is consistent with a major contribution from the resonance form for compound 6 shown above. The C1–N1 (1.3109 (13) Å) and C4–N4 (1.3023 (13) Å) bonds are significantly shorter than C12–N1 (1.3817 (13) Å) and C6–N2 (1.3842 (13) Å) for the same reason. An intramolecular N3–H1n···N4 hydrogen bond (H···N = 2.149 (13) Å, N–H···N = 113.4 (11)°) completes the structure. In the extended structure of compound 6, various aromatic π–π stacking interactions occur, the shortest centroid–centroid separation being 3.7198 (6) Å for the N1 and C2 rings. The structure of compound 6 (Twomey’s method) is almost identical (see experimental section).
It was then realised that this compound is known and had been reported by Twomey and co-workers in 1958.15 This is almost a century since it was first made and left unknown as an industrial synthesis. Twomey’s method of synthesis involves the oxidation of 2-aminodiphenylamine hydrochloride with FeCl3 forming both compounds 4 and 5 followed by heating with aniline, shown in the ‘Introduction’ section (Schemes 1 and 2). It was considered that the complex molecule 6 might form in both processes via intermediates 3–5. Intermediates 4 and 5 were therefore prepared and fully characterised because the early work reported no NMR data. An X-ray single-crystal structure determination was obtained for compound 4 (Figure 2) and a second crystal structure data set was obtained for compound 6 prepared by this different method: refinement showed no significant differences. This provided further proof that product 6 was made by both methods. This is helpful, given the controversy between Kehrmann and Fischer over the structure of these products.
The molecular structure of compound 4 showing 50% displacement ellipsoids.
Compound 4 crystallises with two molecules in the asymmetric unit (Figure 2). In the C1 molecule, the dihedral angle between the C1–C3/C10–C12 and C4–C9 benzene rings is 2.4 (1)° and the pendant C13–C18 and C19–C24 rings are twisted from the C1–C12/N2/N3 fused skeleton (r.m.s. deviation = 0.021 Å) by 71.62 (4) and 80.74 (3)°, respectively. Equivalent data for the corresponding units in the C25 molecule are 2.43 (6)°, 0.025 Å, 81.51 (4)° and 80.54 (4)°, respectively. The C1–C2 and C10–C11 bond lengths (mean = 1.367 Å) are much shorter than the mean of other four bonds in that ring (1.452 Å) again indicating that the resonance form of compound 4 shown above is the major contributor and the same bond length distribution is seen in the C25 molecule. In the crystal, the molecules are linked into a dimer by a pair of N–H···N links (H···N = 2.34 (2) and 2.43 (2) Å; N–H···N = 152.4 (16) and 133 (2)° for N1 and N5, respectively) and the same H atoms also form intramolecular links (H···N = 2.356 (18) and 2.21 (2) Å; N–H···N = 101.2 (14) and 119 (2)° for N1 and N5, respectively). The extended structure of compound 4 features various C–H···π and aromatic π–π stacking interactions.
Although we have not isolated intermediates 3–5 by repeating the Holliday reaction, it seems likely that this is the pathway by which compound 6 forms with hot nitrobenzene acting as a mild oxidant in place of FeCl3 or p-benzoquinone. Scheme 4 is a proposal only for how 2-aminodiphenylamine 3 might be formed. There is literature precedent for the formation of compounds 11 and 13 with these reagents and basic catalysis.26,27 The difficult step is the reduction of compounds 11–13 to form compound 3. However, traces of compound 3 might have been formed by this or by a similar mechanism and this may be the reason why Holliday’s synthesis gave low yields of products and was not commercialised. The dimerisation of compound 3 is a good reaction as previously studied.15
A proposed low-yielding route for making 2-aminodiphenylamine 3 from nitrobenzene and aniline.
There was also competition from other red dyes such as rosaniline or fuchsin28–32 and alizarin,33,34 which were made on a large tonnage scale per year.
Conclusion
The literature chemistry on 2-aminodiphenylamine 3 is summarised and the early controversy between Kehrmann and Fischer noted.1–10 Twomey’s group helped to resolve this dispute by showing that the two products in question both formed from the oxidation of 2-aminodiphenylamine 3,15 but further spectroscopic data are reported here including NMR on the three key products 4–6 and two X-ray single-crystal structure determinations for compounds 4 and 6. The work in Holliday’s patent23 of 1865 has been repeated, leading to the isolation of a red dye 9-phenyl-3-anilinophenazone-2-anil 6. No characterisation data was reported in Holliday’s patent. This same red dye was prepared by Twomey’s research group in 1958, nearly a century later, by the FeCl3 or p-benzoquinone oxidation of 2-aminodiphenylamine hydrochloride and then heating the intermediate products with aniline.15 It seems likely that the red dye made by Holliday’s method, from heating aniline hydrochloride with nitrobenzene, is formed from in situ made 2-aminodiphenylamine followed by oxidation with hot nitrobenzene. Some suggestions are considered as to how it might be formed. This stage of the synthesis is the difficult step and accounts for the low yield of red dye in the Holliday method and the presumed failure to commercialise it as well as the competition from other red dyes in the marketplace.
Experimental
Infrared (IR) spectra were recorded on a diamond-attenuated total reflection (ATR) Fourier transform infrared (FTIR) spectrometer. Ultraviolet (UV) spectra were recorded using a Perkin Elmer Lambda 25 UV-Vis spectrometer with EtOH as the solvent. The term sh means shoulder. 1H NMR and 13C NMR spectra were recorded at 400 and 100.5 MHz, respectively, using a Varian 400 spectrometer. Chemical shifts, δ, are given in ppm and measured by comparison with the residual solvent. Coupling constants, J, are given in Hz. High-resolution mass spectra were obtained at the University of Wales, Swansea, using an Atmospheric Solids Analysis Probe (ASAP) (positive mode) instrument: Xevo G2-S ASAP. Melting points were determined on a Kofler hot stage microscope.
9-Phenyl-3-anilinophenazone-2-anil6 (Method 1) Aniline hydrochloride (1.5 g, 11.6 mmol) and nitrobenzene (3.0 g, 24.4 mmol) were mixed and heated at 200 °C forming a purple melt. If a melt does not form, more nitrobenzene is added, but if too much is added, it washes products through the sinter when the reaction is worked up. Aniline hydrochloride sublimes under these conditions. After 1 h, the melt was cooled and mixed with dilute aq 1M HCl (50 mL) and filtered. The sinter No 4 was washed with dichloromethane (DCM) (50 mL) and the filtrate collected. A blue product on the sinter was also collected by washing with acetone (50 mL) or methanol (50 mL). The fractions were mixed and evaporated to dryness. The mixture was purified by chromatography on flash silica. Elution with Et2O gave the title compound (12 mg, 0.2%) as red crystals, m.p. 235–237 °C (from DCM: light petroleum ether). λmax (EtOH) nm−1 360 (log ε 3.5) and 272 (3.8); λmax (diamond)(cm−1) 2919m, 1594m, 1510s, 1372m, 1247s, 1150m, 1072m, 1026m, 812w, 747s, 694s, 604m and 510m; δH (400 MHz; CDCl3) 5.33 (1H, s), 6.47 (1H, d, J = 8.0), 6.73 (2H, d, J = 8.0), 6.85 (1H, t, J = 8.0 and 8.0), 6.98–7.20 (8H, m), 7.28–7.37 (4H, m), 7.42 (1H, d, J = 8.0), 7.49 (2H, t, J = 8.0 and 8.0), 7.69 (1H, d, J = 8.0) and 8.40 (1H, s, br); δC (100.1 MHz; CDCl3) 91.0, 99.7, 114.4, 121.3, 122.0, 123.1, 123.2, 123.7, 127.7, 128.4, 128.5, 128.7, 129.4, 129.6, 130.7, 131.4, 135.2, 136.0, 137.2, 139.8, 144.0, 150.7 and 152.5 (one resonance is overlapping); m/z (Orbitrap ASAP) 439.1929 (M+ + H, 100%) C30H23N4 requires 439.1923; the method was not optimised. Then, 20% aqNH3/MeOH eluted a polar blue compound 10 (8 mg, 0.2%) λmax (EtOH) nm−1 574 (log ε 3.5) and 284 (3.7); λmax (diamond) (cm−1) 3140s, 3040s, 1590s, 1490s, 1301s, 1248s, 1176s, 1152s, 1130s, 1074s, 825w, 750s, 695s, 610s and 495s; m/z (Orbitrap ASAP) 454.2036 (M+ + H, 100%) C30H24N5 requires 454.2032.
2-Amino-3:5-dihydro-5-phenyl-3-phenyliminophenazine4and 2-Anilino-3:5-dihydro-3-imino-5-phenylphenazine5 2-Aminodiphenylamine hydrochloride (500 mg, 2.27 mmol) in water (30 mL) was treated with Fe(III)Cl3.6H2O (1.38 g, 5.1 mmol) and stirred at room temperature for 3 h. The mixture was filtered. Dilute KOH (0.1 M) was passed through the precipitate in the sinter. The filtrate turned grey when it was neutralised but contained no red products. The red products were purified by chromatography on flash silica. The mixture was loaded onto the column with DCM. Elution with Et2O gave 2-amino-3:5-dihydro-5-phenyl-3-phenyliminophenazine (49 mg, 10%) as red crystals, m.p. 257–259 °C (from DCM: light petroleum ether). λmax (EtOH) nm−1 452 (log ε 3.7) and 270 (3.9); λmax (diamond)(cm−1) 3350w,1534s, 1493s, 1461s, 1353m, 1318m, 1246m, 1208m, 1067m, 1020m, 955w, 909w, 845m, 813s, 761m, 741s, 692s, 609m, 584m, 499m and 437s; δH (400 MHz; CDCl3) 5.35 (1H, s), 5.38–5.52 (2H, s, br, NH), 6.50 (2H, t, J = 8.0 and 8.0), 6.74 (2H, d, J = 8.0), 6.86 (1H, t, J = 8.0 and 8.0), 7.05–7.17 (6H, m), 7.41 (1H, t, J = 8.0 and 8.0), 7.48 (2H, t, J = 8.0 and 8.0) and 7.72 (1H, d, J = 8.0); δH (400 MHz; DMF D7) 5.30 (1H, s), 6.38 (1H, d, J = 8.0), 6.48 (1H, s), 6.53–6.58 (2H, s, br, NH), 6.65 (2H, d, J = 8.0),6.78 (1H, t, J = 8.0 and 8.0), 7.01–7.13 (4H, m), 7.33 (2H, d, J = 8.0), 7.43 (1H, t, J = 8.0 and 8.0), 7.51–7.59 (3H, m); δC (100.1 MHz; DMF D7) 90.6, 99.5, 114.5, 121.2, 122.9, 123.0, 127.3, 127.8, 128.7, 128.8, 129.6, 131.0, 134.8, 136.0, 137.6, 150.1, 150.4, 151.2 and 152.7 (one resonance is overlapping); m/z (Orbitrap ASAP) 363.1609 (M+ + H, 100%) C24H19N4 requires 363.1610. Elution with 20% aqNH3/MeOH gave 2-anilino-3:5-dihydro-3-imino-5-phenylphenazine (218 mg, 44%) as dark amorphous material, m.p. 200–202 °C. λmax (EtOH) nm−1 469 (log ε 4.2) and 278 (4.3); λmax (diamond)(cm−1) 1564s, 1490s, 1463s, 1315s, 1294s, 1243s, 1024m, 951w, 836m, 742s, 691s, 608s, 591s and 502s; δH (400 MHz; CDCl3) 4.40–5.10 (2H, s, br, NH), 5.51 (1H, s), 6.64 (1H, d, J = 8.0), 7.06–7.16 (2H, m), 7.22–7.45 (8H, m), 7.64–7.79 (3H, m) and 7.84 (1H, d, J = 8.0); δC (100.1 MHz; CDCl3) 96.2, 100.9, 115.0, 122.2, 123.9, 124.1, 128.4, 128.6, 128.8, 129.4, 130.2, 130.9, 131.3, 134.3, 136.3, 137.0, 139.5, 143.1, 149.8 and 160.1; m/z (Orbitrap ASAP) 363.1605 (M + H, 100%) C24H19N4 requires 363.1610.
9-Phenyl-3-anilinophenazone-2-anil6 (Method 2) A mixture of 2-amino-3:5-dihydro-5-phenyl-3-phenyliminophenazine4 and 2-anilino-3:5-dihydro-3-imino-5-phenylphenazine5 (30 mg, 0.083 mmol) was heated in aniline (20 mL) with aniline hydrochloride (0.5 g, 3.8 mmol) for 15 min. The cooled mixtures were poured into dilute aq HCl (10 mL c. HCl diluted in 150-mL H2O), extracted with DCM (100 mL) and dried over MgSO4. The product was purified by chromatography on flash silica. Gradient elution with Et2O/DCM (10/90), then (30/70), then (50/50) eluted the title compound (16 mg, 42%), m.p. 235–237 °C with identical spectroscopic properties to the material made by Holliday in Method 1. The proton and carbon 13 NMR data of this second batch of material are reproduced in the ‘Supplemental material’ section.
Single-crystal diffraction
The crystal structures were established using intensity data collected on a Rigaku CCD diffractometer at T = 100 K. The structures were routinely solved by dual-space methods using SHELXT,35 and the structural models were completed and optimised by refinement against |F|2 with SHELXL-2018.36 The N-bound hydrogen atoms were located in difference maps, and their positions were freely refined. The C-bound hydrogen atoms were placed in idealised locations (C–H = 0.95 Å) and refined as riding atoms. The constraint Uiso(H) = 1.2Ueq(carrier) was applied in all cases. Full details of the structures and refinements are available in the deposited cifs.
Crystal data for compound 6 (Holliday) C30H22N4 (red rod 0.32 × 0.06 × 0.04 mm, recrystallised from DCM/light petrol 40–60 solution), Mr = 438.51, triclinic, space group P (No. 2), a = 9.9976(2) Å, b = 10.6030 (2) Å, c = 11.6145 (2) Å, α = 108.439 (2)°, β = 104.957 (2)°, γ = 92.334 (2)°, V = 1118.31 (4) Å3, Z = 2, T = 100 K, Cu Kα radiation, λ = 1.54178 Å, μ = 0.609 mm−1, ρcalc = 1.302 g cm−3, 64,579 reflections measured (8.4 ⩽ 2θ ⩽ 153.7°), 4528 unique (RInt = 0.025), R(F) = 0.034 (4149 reflections with I > 2σ(I)), wR(F2) = 0.096 (all data), Δρmin,max (e Å−3) = −0.21, +0.28, CCDC deposition number 2244635.
Crystal data for compound 6 (Twomey) C30H22N4 (red block 0.17 × 0.15 × 0.08 mm, recrystallised from DCM/light petrol 40–60 solution), Mr = 438.51, triclinic, space group P (No. 2), a = 9.9857 (2) Å, b = 10.6012 (2) Å, c = 11.6189 (2) Å, α = 108.4660 (10)°, β = 104.9320 (10)°, γ = 92.2640 (10)°, V = 1117.37 (4) Å3, Z = 2, T = 100 K, Mo Kα radiation, λ = 0.71073 Å, μ = 0.078 mm−1, ρcalc = 1.303 g cm−3, 64,353 reflections measured (4.1 ⩽ 2θ ⩽ 67.4°), 8143 unique (RInt = 0.030), R(F) = 0.047 (6908 reflections with I > 2σ(I)), wR(F2) = 0.136 (all data), Δρmin,max (e Å−3) = −0.21, +0.57, CCDC deposition number 2245056.
Crystal data for compound 4 C24H18N4 (light yellow prism, 0.08 × 0.04 × 0.03 mm, recrystallised from DCM/light petrol 40–60 solution), Mr = 362.42, triclinic, space group P (No. 2), a = 11.1873 (3) Å, b = 13.3083 (4) Å, c = 13.7177 (3) Å, α = 113.543 (2)°, β = 92.690 (2)°, γ = 102.281 (2)°, V = 1809.55 (9) Å3, Z = 4, T = 100 K, Cu Kα radiation, λ = 1.54178 Å, μ = 0.633 mm−1, ρcalc = 1.330 g cm−3, 56,344 reflections measured (7.1 ⩽ 2θ ⩽ 153.6°), 7296 unique (RInt = 0.034), R(F) = 0.044 (6078 reflections with I > 2σ(I)), wR(F2) = 0.126 (all data), Δρmin,max (e Å−3) = −0.25, +0.64, CCDC deposition number 2244634.
Supplemental Material
sj-docx-1-chl-10.1177_17475198231175602 – Supplemental material for Uncovering John Holliday’s industrial dye synthesis patented in 1865
Supplemental material, sj-docx-1-chl-10.1177_17475198231175602 for Uncovering John Holliday’s industrial dye synthesis patented in 1865 by Michael John Plater and William TA Harrison in Journal of Chemical Research
Footnotes
Acknowledgements
The authors thank the UK Engineering and Physical Sciences Research Council (EPSRC) National Mass Spectrometry Service Centre for mass spectrometric data and the UK National Crystallography Centre (University of Southampton) for the X-ray data collections. Data sets were obtained free of charge from the National Crystallography Centre, Southampton University.
Author contributions
M.J.P. performed all synthesis and obtained the characterisation data and W.T.A.H. solved the crystallographic data sets.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
ORCID iD
Michael John Plater
Supplemental material
Supplemental material for this article is available online.
BarryVCBeltonJGConaltyML, et al. Nature1948; 162: 622–623.
12.
BarryVCBeltonJG. Proc R Ir Acad Sect B Biol Geol Chem Sci1953; 55: 149–156.
13.
BarryVCBeltonJGChambersJF, et al. Proc R Ir Acad Sect B Biol Geol Chem Sci1953; 55: 157–164.
14.
BarryVCBeltonJGO’SullivanJF, et al. J Chem Soc (Resumed)1956: 888–893.
15.
BarryVCBeltonJGO’SullivanJF, et al. J Chem Soc (Resumed)1958: 859–863.
16.
BeltonJGO’CallaghanCNTwomeyD. Proc R Ir Acad Sect B Biol Geol Chem Sci1961; 62: 9–14.
17.
BarryVCBeltonJGConaltyML, et al. Proc R Ir Acad Sect B Biol Geol Chem Sci1970; 70: 179–194.
18.
BarryVCBeltonJGO’SullivanJF, et al. J Chem Soc (Resumed)1959: 3217–3223.
19.
BarryVCBeltonJGO’SullivanJF, et al. J Chem Soc (Resumed)1956: 896–899.
20.
BarryVCBeltonJGO’SullivanJF, et al. J Chem Soc (Resumed)1956: 3347–3350.
21.
BarryVCBeltonJGO’SullivanJF, et al. J Chem Soc (Resumed)1956: 893–895.
22.
BarryVCBeltonJGO’SullivanJF, et al. J Chem Soc (Resumed)1958: 4495–4498.
23.
HollidayJ. Patent application 2564, GB, 1865.
24.
Patents for inventions. Abridgements of specifications relating to bleaching dyeing and printing calico and other fabrics and yarns. The manufacture of rollers, engraving, the preparation of drugs and other processes. Pub Eyre and Spottiswoode Part I 1-770; Part II 1-662; Part III 1-349; Part IV 1-316.
25.
FoxMR. Dye makers of Great Britain. A history of chemists, companies, products and changes 1856–1976. Manchester: Imperial Chemical Industries PLC, 1987.
26.
BeskaETomanPFiedlerK, et al. Patent 6388136 B1, USA, 2002.
27.
PlaterMJ. Molbank2022; 2022: M1406.
28.
BroomanRA. Patent application 921, GB, 1859.
29.
PerkinWH. Patent application 2492, GB, 1859.
30.
MedlockH. Patent application 126, GB, 1860.
31.
NicholsonEC. Patent application 184, GB, 1860.
32.
De LaireGGirardC. Patent application 1300, GB, 1860.
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.