
Abstract
[2.2]-Paracyclophane derivatives have been promising platforms for applications in biology and materials science. To access pure derivatives thereof, a safe and non-toxic synthetic method for 4-n-propyl-[2.2]-paracyclophane is developed via a microwave-assisted and NH2NH2/KOH reduction route. We introduce microwave-assisted acylation for a synthesis that successfully improves the yield and reduces the reaction time. Notably, an exploration of the length and number of the alkyl chains on the [2.2]-paracyclophane ring did not significantly affect the outcome of this reaction. We synthesized 4-n-butyl-[2.2]-paracyclophane and 4,12-dipropyl-[2.2]-paracyclophane via our protocol with high yields. The regioselectivity of the second electrophilic substitution on 4-n-propyl-[2.2]-paracyclophane occurs at the para position of the less substituted phenyl ring.

Introduction
Parylene thin film, which is polymerized by chemical vapor deposition of [2.2]-paracyclophane and its substituents, is one of the most advanced coating materials in the world.1–5 It is widely used because of its excellent performance characteristics, such as electrical insulation, 6 compact structure, good heat resistance, 7 mildew and salt spray resistance, 8 good biocompatibility, etc. It also plays important roles in many fields such as semiconductors, integrated circuits, micro-electro-mechanical systems, sensors, medical instruments, and for cultural relic protection.9–12
Current studies have shown that performance optimization and transformation can be achieved by attaching different substituents on the [2.2]-paracyclophane, thus adapting it to more application scenarios. For example, chlorine-substituted [2.2]-paracyclophane has good electrical and physical properties, which can effectively reduce damage caused by corrosive gases and prolong its life. 13 Likewise, 4-acetyl-[2.2]-paracyclophane is a promising platform for studies of planar chirality and through-space electronic communications in π-stacked molecular systems. The literature shows that [2.2]-paracyclophane containing alkyl chains can effectively enhance the thin film stretching rate of parylene film and reduce its solvent resistance. 14 Such materials are expected to be applied in more precise instruments as coatings. For example, poly (n-propyl-p-xylylene) has a tensile strength at a breaking point of up to 380%, which is 10% to 15% of that of parylene N, the base member of this series. The crystallization point of the film prepared from 4-n-butyl-[2.2]-paracyclophane by the Chemical Vapor Deposition (CVD) method is 25 °C, and its ability to undergo high fracture elongation can meet application needs in medical telescopic lenses.15,16 However, the crystallinity of the polymer decreases compared with other films, while, its solvent resistance is also greatly reduced. 17
Since its introduction in the 1960s, parylene film has received unprecedented attention from many countries, including the United States, and as a result, it has replaced the original tri-proof material to a certain extent and is more often used in the field of coating. 18 There are already commercially available film precursors of parylene N, parylene C, parylene D, and parylene HT ( Figure 1 ).19,20 But there are still technical obstacles to research on precursors for alkylated parylene products with unique advantages. Yeh and Gorham 21 reported the synthesis of 4-ethyl-[2.2]-paracyclophane in an 85% yield via a Friedel Crafts alkylation on [2.2]-paracyclophane using bromoethane in the presence of aluminum chloride. However, this method is not suitable for the synthesis of straight alkanes with three or more carbons because long-chain alkanes can easily rearrange via more stable carbocations in the presence of Lewis acids ( Scheme 1 ). 22 Bier et al. 23 reported the synthesis of 4,16-diethyl-[2.2]-paracyclophane in an 80% yield via a Kumada coupling reaction carried out on 4,16-dichloro-[2.2]-paracyclophane using a Grignard reagent and 1,3-bis(diphenylphosphino)propane nickel chloride as the catalyst. However, 1,3-bis(diphenylphosphino)propane nickel chloride is expensive and the use and preparation of Grignard reagents are environmentally stringent providing obstacles to the synthesis on a large scale ( Scheme 2 ). Therefore, a more convenient, low-cost, and green protocol for the synthesis of alkyl-substituted [2.2]-paracyclophanes is required.
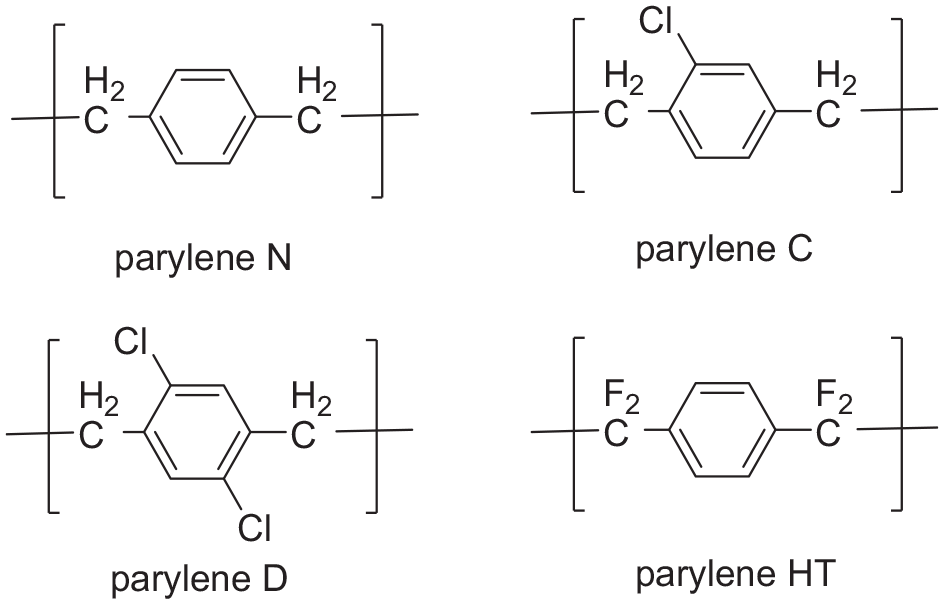
Chemical structures of parylene N, parylene C, parylene D, and parylene HT.

The previous work reported by Gorham et al.

The previous work reported by Bier et al.
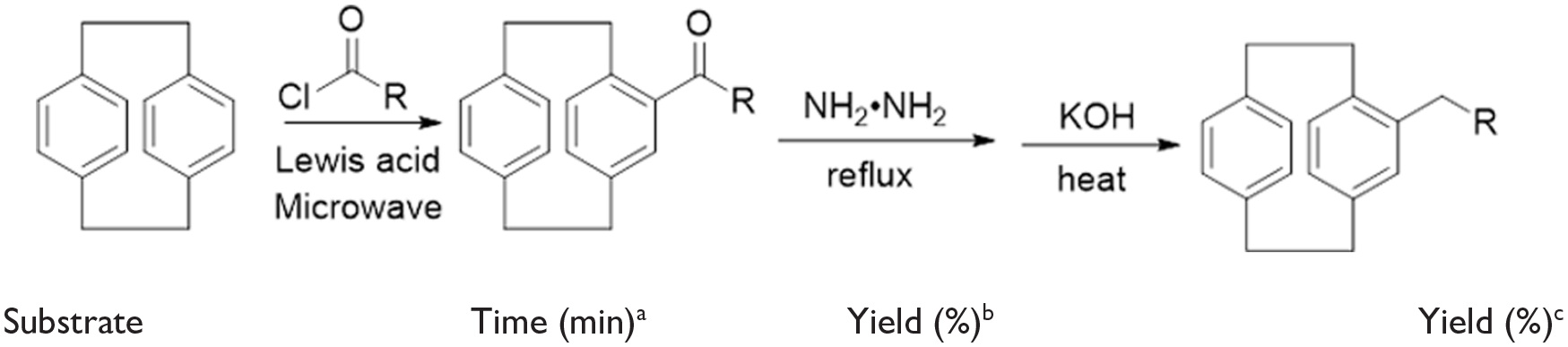
Herein, we report a new microwave-assisted protocol for the synthesis of long-chain alkyl substituted [2.2]-paracyclophanes with a useful procedure for the reduction of aldehyde and ketone containing side-chains on the [2.2]-paracyclophane framework. This method avoids long reaction times and low yields. Overall, it is a green, efficient, and sustainable route that employs simple equipment, a safe operation procedure, short reaction times, and has prospects for many applications.
Results and discussion
We started our investigations on the acylation of [2.2]-paracyclophane (
Optimization of the reaction conditions for the preparation of 4-propionyl-[2.2]-paracyclophane (


Isolated yields of products based on
Interestingly, we never observed the formation of a diacylated-[2.2]-paracyclophane during these reactions, despite the propionyl chloride and the Lewis acid being present in excess. In contrast, acylation reactions of simple alkyl derivatives of benzene do not stop at the monoacyl substitution stage. The lack of formation of diacylated-[2.2]-paracyclophane is because the initial arenium ion formed is stabilized through favorable trans-ring interactions. 21 Therefore, it is impractical to obtain the disubstituted [2.2]-paracyclophane by a one-pot process.
Considering our overarching goal of developing a green protocol for the process we reckoned that there were still problems with our procedure such as high energy consumption, long reaction times, and moderate yields, which made it uneconomic. We have, therefore, explored microwave-assisted methods to try and solve the above problems. Microwave-assisted synthesis has become commonplace these days as a means of reducing reaction times and thus increasing the efficiency of the reaction from an energy consumption point of view.24,25 We have, therefore, explored microwave-assisted reaction conditions and the results are listed in
Table 2
. A mixture of [2.2]-paracyclophane, AlCl3, and propionyl chloride was irradiated three times in an open microwave reactor, then we attempted to synthesize
Microwave-assisted synthesis of
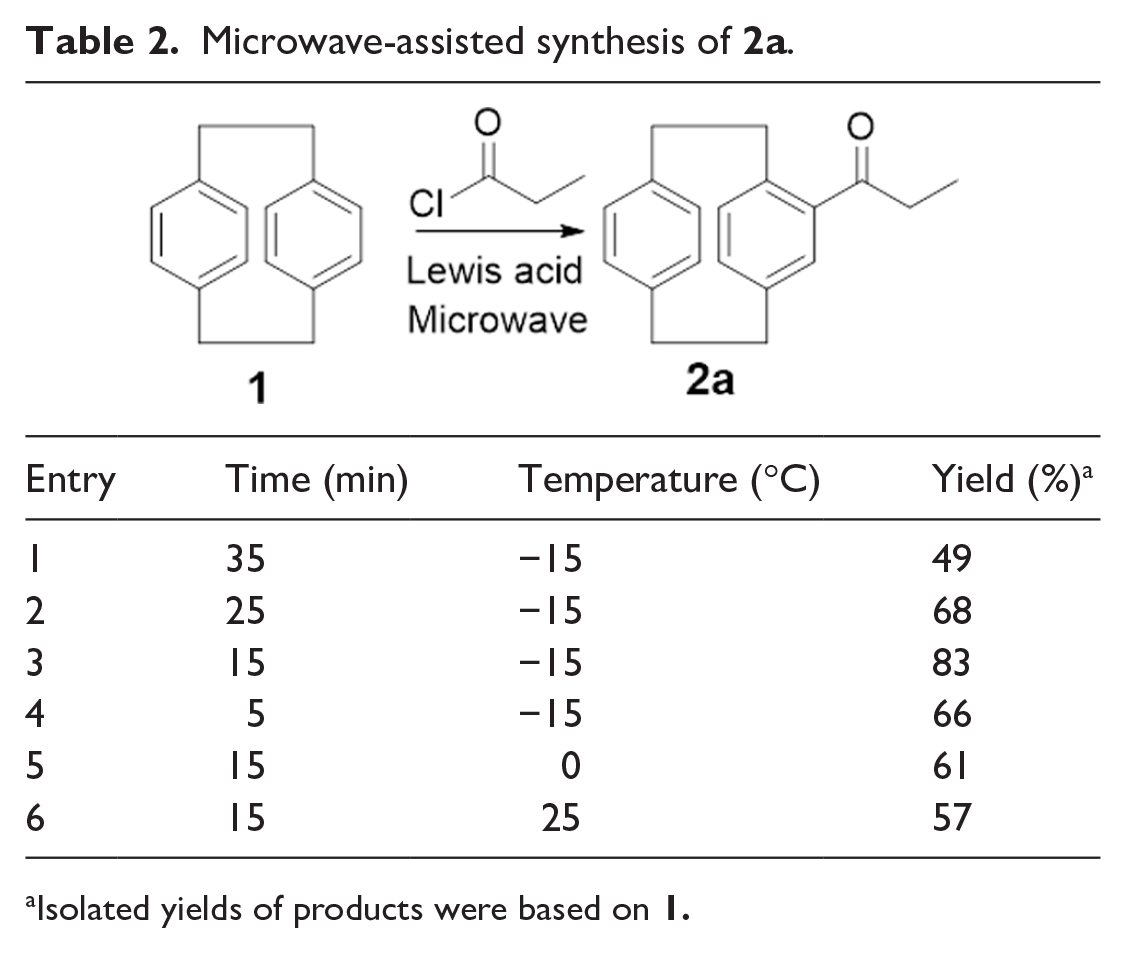

Isolated yields of products were based on
Next we set out to explore the reduction of compound
Attempted hydrogenation of
Optimization of the reaction conditions for the synthesis of 4-n-propyl-[2.2]-paracyclophane (


Catalytic hydrogenation experiment.
NaBH4/AlCl3 system.
Isolated yields of products based on
We investigated the effect of the temperature on the yield of this reduction. Higher reaction temperatures, up to 225 °C, helped to shorten the reaction time and increased the yield (
Table 3
, entry 4). However, the yield decreased and product charring occurred when the reduction temperature was too high, and it is thus desirable to monitor the reaction temperature carefully (
Table 3
, entry 5). Hydrazine and potassium hydroxide play a role in the generation of the hydrazone intermediate. At high temperatures, it is likely that the hydrazine may undergo decomposition and thus a slight excess of this reagent is desirable. Whereas caustics are relatively stable at high temperatures, given the cost and environmental impact of these reagents, it is desirable to keep the ratio of the caustic reagent to substrate low (
Table 3
, entries 6–8). The reflux time has a minor effect with longer times leading to a decrease in the yield of compound
With optimum reaction conditions in place, (
Table 2
, entry 3 and
Table 3
entry 8), we proceeded to see whether the length of the alkyl chain on the [2.2]-paracyclophane ring affected the outcome of this reaction. First, we studied the introduction of alkyl groups with longer carbon chains as shown in
Table 4
. 4-Butyryl-[2.2]-paracyclophane (
Synthesis of monoalkyl-substituted [2.2]-paracyclophanes.

Reaction time for the acylation.
Isolated yields of products based on
Isolated yields of products based on
Having optimized the reaction conditions for the acylation step and the subsequent reduction step for the synthesis of
Acylation of 4-n-propyl-[2.2]-paracyclophane
Therefore, as shown in
Table 5
, 4-propyl-12-propionyl-[2.2]-paracyclophane (
Synthesis of dialkyl-substituted [2.2]-paracyclophanes.


Reaction time for the acylation.
Isolated yield of product based on
Isolated yield of product based on
Conclusion
We have developed a rapid, efficient and highly selective microwave-assisted method for the synthesis of acyl-substituted [2.2]-paracyclophanes, and have identified an efficient method for the reduction of these ketones for the synthesis of alkyl-substituted [2.2]-paracyclophanes. This work demonstrates the feasibility of microwave-assisted synthesis in the acylation of [2.2]-paracyclophanes and establishes the high efficiency of the NH2NH2/KOH reduction for the reduction of long chain acyl groups on the [2.2]-paracyclophane framework. This protocol greatly reduces the reaction time, reduces the energy consumption, uses mild reagents with low toxicity and mild reagents, and provides good yields. It provides a new method for the synthesis of parylene precursors with long-chain alkyl substituents.
Experimental section
General information
Hydrazine hydrate(80%), ethylene glycol, methylene chloride, potassium hydroxide, chloroform, 1,1,2,2-tetrachloroethane, titanium tetrachloride, sodium bicarbonate, hydrochloric acid, petroleum ether (60~90 °C), and ethyl acetate were purchased from Sinopharm Chemical Reagent Co., Ltd. Anhydrous aluminum chloride, anhydrous magnesium sulfate, propionyl chloride, Raney nickel, and ferric chloride were obtained from Aladdin (Shanghai, China). Chloroform-d (D, 99.8%) was purchased from Adamas Reagent Co., Ltd. [2.2]-Paracyclophane was provided by Suzhou Kaida Biomedical Technology Co., Ltd. Nitrogen (N2, 99.99%) was purchased from Nanjing Chuangda special gas Co., Deionized water was self-prepared in our laboratory. All reagents were of analytical purity and were used without further purification.
Mcrowave-assisted reactions were carried out in an open-vessel system using a XH-100B microwave reactor (Beijing XiangHu, China) at a constant power (600 W). NMR spectra were measured on Bruker Avance III 400 MHz or Bruker Ascend 600 MHz spectrometers. Melting points were determined using a Beijing TaiKe X-4 melting point apparatus and are uncorrected. MS data were recorded on Agilent 7250.
Acyl-substituted [2.2]-paracyclophane; general procedure
[2.2]-Paracyclophane (2.09 g, 10 mmol) was dissolved in dichloromethane (100 mL) under an atmosphere of nitrogen with stirring in cryogenic coolant circulation pumps. Next, anhydrous aluminum chloride (2.13 g, 16 mmol) and propionyl chloride (1.47 g, 16 mmol) were added to the reaction system. The mixture was stirred at −15 °C for 40 min. Then, the reaction was quenched by the addition of ice-cold hydrochloric acid (2 mol/L, 100 mL). The organic phase was separated and washed several times with sodium bicarbonate and water until neutral. The organic fraction was dried, and the CH2Cl2 was removed by evaporation under vacuum to obtain a pale yellow solid. The pale yellow solid was purified by column chromatography (SiO2, ethyl acetate/petroleum ether 60~90 °C, 1:15 v/v) to obtain a white solid powder. The product was identified by comparison with literature data. 28
Microwave-assisted synthesis of acyl-substituted [2.2] paracyclophane
A mixture of [2.2]-paracyclophane (2.09 g, 10 mmol), AlCl3 (2.13 g,16 mmol) and propionyl chloride (1.47 g, 16 mmol) was irradiated three times in an open microwave reactor for 5 min each. The reaction was then continued at −15 °C for 15 min. following to the procedure described above.
Alkyl-substituted [2.2]-paracyclophane; general procedure
4-Propionyl-[2.2]-paracyclophane (1.32 g, 5 mmol), potassium hydroxide (0.84 g, 15 mmol), and diethylene glycol (30 mL) were stirred in a 100 mL three-neck flask until the compounds had completely dissolved. 80% Hydrazine hydrate (1.87 g, 30 mmol) was added dropwise to the mixture, and the mixture was heated at 180 °C under reflux for 2 h with stirring. The reflux condenser was carefully removed for a moment to evaporate the excess hydrazine hydrate and water, then the reaction temperature of the mixture was raised to 225 °C and stirring was continued for 3 h. After cooling, the mixture was adjusted to neutral by adding dilute hydrochloric acid and extracted with CH2Cl2 (3×15 mL) to obtain the organic phase. The combined organic extract was dried over MgSO4 and concentrated under vacuum to obtain the white solid. The white solid was purified by column chromatography (SiO2, ethyl acetate/petroleum ether 60~90 °C, 1:50 v/v) to obtain a white solid powder which is the product. The product was identified by comparison with literature data.23,29
Dialkyl-substituted [2.2]-paracyclophane; general procedure
A mixture of compound
Compound
[2.2]-paracyclophane (1 )
White solid. m.p. 281–283 °C .1H NMR (600 MHz, CDCl3): δ = 6.41 (s, 8H), 3.01 (s, 8H). 13C NMR (150 MHz, CDCl3): δ =139.2, 132.8, 36.1.
4-propionyl-[2.2]-paracyclophane (2a )
White solid powder. m.p. 263 °C. 1H NMR (600 MHz, CDCl3): δ = 6.89 (s, 1H), 6.65–6.34 (m, 6H), 3.88 (t, J = 11.2 Hz, 1H), 3.22–2.68 (m, 9H), 1.19 (t, J =7.3 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ = 203.4, 141.1, 140.2, 139.6, 139.0, 137.7, 136.3, 136.0, 133.1, 132.9, 132.7, 132.0, 131.1, 35.9, 35.1, 35.1, 35.0, 33.6, 8.6.
4-n-propyl-[2.2]-paracyclophane (2b )
White solid powder. m.p. 103 °C–104 °C. 1H NMR (600 MHz, CDCl3): δ = 6.69 (dd, J = 7.8, 1.6 Hz, 1H), 6.53 (dd, J = 7.8, 1.6 Hz, 1H), 6.47 (dd, J = 7.8, 1.4 Hz, 1H), 6.41 (d, J = 7.7 Hz, 3H), 6.14 (s, 1H), 3.41–3.32 (m, 1H), 3.16–2.93 (m, 6H), 2.84–2.76 (m, 1H), 2.63–2.54 (m, 1H), 2.30–2.20 (m, 1H), 1.48 (dd, J = 15.0, 7.5 Hz, 2H), 0.91 (t, J = 7.3 Hz, 3H), 13C NMR (150 MHz, CDCl3): δ = 142.1, 139.6, 139.6, 139.5, 137.6, 134.7, 134.7, 133.4, 133.2, 132.2, 130.2, 129.0, 36.7, 35.5, 35.2, 34.4, 33.7, 23.8, 14.3. MS (EI): m/z [M ]+ calcd for C19H22: 250.17; found: 250.20.
4-butyryl-[2.2]-paracyclophane (3a )
White solid powder. m.p. >300 °C. 1H NMR (600 MHz, CDCl3): δ = 6.81 (s, 1H), 6.57–6.27 (m, 6H), 3.79 (ddd, J = 12.3, 10.0, 1.9 Hz, 1H), 3.18–3.11 (m, 1H), 3.14–3.06 (m, 2H), 3.09–2.99 (m, 1H), 2.99–2.90 (m, 2H), 2.81–2.69 (m, 2H), 2.58 (ddd, J = 16.4, 8.2, 6.2 Hz, 1H), 1.73–1.56 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3): δ = 203.09, 141.23, 140.31, 139.71, 139.16, 137.95, 136.32, 136.08, 133.22, 132.92, 132.82, 132.17, 131.20, 42.59, 35.96, 35.20, 35.18, 35.10, 18.07, 13.94.
4-n-butyl-[2.2]-paracyclophane (3b )
White solid powder. m.p. 67 °C–69 °C (literature value: 65–68 °C). 1H NMR (600 MHz, CDCl3) δ = 6.61 (dd, J = 7.8, 1.9 Hz, 1H), 6.44 (d, J = 7.8 Hz, 1H), 6.38 (m, 1H), 6.36–6.29 (m, 3H), 6.05 (d, J = 7.8 Hz, 1H), 3.31–3.23 (m, 1H), 3.09–2.82 (m, 6H), 2.76–2.66 (m, 1H), 2.54–2.47 (m, 1H), 2.22–2.14 (m, 1H), 1.40–1.30 (m, 2H), 1.27–1.20 (m, 2H), 0.82 (t, J = 7.3 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ = 142.17, 139.55, 139.45, 139.41, 137.40, 134.58, 134.53, 133.26, 133.11, 132.13, 130.09, 128.84, 35.39, 35.08, 34.29, 34.12, 33.54, 32.69, 22.70, 14.01. MS (EI): m/z [M ]+ calcd for C20H24: 264.19; found: 264.19.
4-propyl-12-propionyl-[2.2]-paracyclophane (4a )
White solid powder. 1H NMR (600 MHz, CDCl3): δ = 6.85 (s, 1H), 6.66 (dd, J = 7.8, 1.8 Hz, 1H), 6.47 (m, 2H), 6.36 (dd, J = 7.9, 1.7 Hz,1H), 6.18 (s, 1H), 3.97–3.89 (m, 1H), 3.44–3.35 (m, 1H), 3.2–3.08 (m, 3H), 3.02–2.85 (m, 2H), 2.81–2.57 (m, 4H), 2.36–2.26 (m, 1H), 1.49–1.33 (m, 2H), 1.19 (t, J = 7.3 Hz, 3H), 0.91 (t, J = 7.3 Hz, 3H). 13C NMR (150 MHz, CDCl3): δ = 203.0, 146.6, 141.6, 140.2, 139.0, 137.7, 137.5, 135.7, 135.1, 132.2, 132.0, 131.5, 129.1, 36.2, 35.8, 34.8, 33.8, 33.6, 33.4, 23.2, 14.1, 8.7.
4, 12-dipropyl-[2.2]-paracyclophane (4b )
Colorless liquid. 1H NMR (600 MHz, CDCl3): δ = 6.74 (d, J = 7.9 Hz, 2H), 6.43 (d, J = 9.4 Hz, 2H), 6.10 (s, 2H), 3.39–3.31 (m, 2H), 3.12 (t, J= 11.6 Hz, 2H), 3.08–3.00 (m, 2H), 2.76–2,66 (m, 2H), 2.63–2.52 (m, 2H), 2.35–2.26 (m, 2H), 1.53 (dd, J = 14.7, 7.3 Hz, 4H), 0.97 (t, J = 7.3 Hz, 6H). 13C NMR (151 MHz, CDCl3): δ = 139.5, 139.4, 137.2, 136.0, 132.4, 129.5, 36.2, 34.0, 33.3, 23.8, 14.4. MS (EI): m/z [M ]+ calcd for C22H28 292.22; found: 292.31.
Supplemental Material
sj-docx-1-chl-10.1177_17475198231164370 – Supplemental material for A microwave-assisted and efficient reduction protocol for the synthesis of alkyl-substituted [2.2]-paracyclophanes
Supplemental material, sj-docx-1-chl-10.1177_17475198231164370 for A microwave-assisted and efficient reduction protocol for the synthesis of alkyl-substituted [2.2]-paracyclophanes by Mengyuan Tian, Lixiang Zhang, Lijing Gao and Guomin Xiao in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the National Key R&D Program of China (Number 2019YFB1504003).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.