
Abstract
This work describes the preparation of manganese-doped mesoporous silica nanospheres via an in situ doping method. The results of scanning electron microscopy and N2 adsorption demonstrate that mesoporous silica possesses a spherical shape, a highly porous structure, a large specific surface area of 922.21 m2 g−1, and a pore volume of 0.257 cm3 g−1. The mesoporous silica nanocarrier is loaded with doxorubicin, and carboxymethyl chitosan encapsulation is performed to prevent doxorubicin leakage. The easy release characteristics of manganese under acidic conditions and the swelling properties of carboxymethyl chitosan endow the drug-loading system with an excellent pH/responsive release property. A cytotoxicity test shows that mesoporous silica nanospheres–doxorubicin–carboxymethyl chitosan had significant biocompatibility and enhanced cytotoxicity, thus revealing mesoporous silica nanospheres–doxorubicin–carboxymethyl chitosan as a promising delivery system.
By in situ doping method, manganese-doped mesoporous silica nanospheres (MMS) were prepared, which were then loaded with doxorubicin (DOX) and coated using a carboxymethyl chitosan (CMCS) to develop a nanocarrier delivery system (MMS-DOX-CMCS). Due to the dissolution characteristics of manganese ions and swelling behavior of carboxymethyl chitosan, the MMS-DOX-CMCS delivery system exhibited a good pH-responsive release.

Introduction
Recently, inorganic nanostructured materials have attracted significant attention due to their physiological stability, easy functionalization, and other unique physiochemical properties. 1 The employment of inorganic nanoparticles as drug delivery systems2–6 has generated intense interest owing to their high biosafety, delivery efficiency, and controllable drug-releasing profiles. Mesoporous silica (SiO2) nanoparticles have several advantages including large specific surface areas, porous structures, good biocompatibility, and modifiable surfaces.7–9 However, notable challenges remain in translating SiO2 from basic research clinical use. For instance, the Si–O–Si skeleton is stable and difficult to degrade. Some strategies have been proposed to improve the biodegradability of SiO2 via reconstructing the framework of silica nanocarriers, such as inorganic–organic hybridization 10 and metal iondoping 1 (e.g. iron, 11 manganese, 12 calcium, 13 magnesium 14 ). Therefore, we introduced manganese (Mn) into the framework of hollow SiO2 nanospheres to form the Si–O–Mn skeleton. It was hoped that the introduction of Mn would significantly change the intrinsic physiochemical properties and biological behavior of silica. In the acidic microenvironment of tumors, the Mn–O bond is broken and the Mn component is released from the silica framework, accelerating cleavage of the Si–O–Si framework. 15
Doxorubicin (DOX; Figure 1) is a chemotherapy medication used to treat various kinds of cancers, including breast cancer 16 and gastric carcinoma. 17 In this paper, DOX as a model drug was encapsulated in the nanocarrier. The therapeutic efficiency of DOX is dependent on its sufficient accumulation within the nucleus of cancer cells. Therefore, after loading DOX, biocompatible carboxymethyl chitosan as the gating switch was constructed on the surface of the loaded Mn-doped SiO2 nanospheres to block DOX. Next, the drug-loading system was characterized by N2 adsorption/desorption, thermogravimetric analysis, Zeta potential analysis, scanning electron microscopy (SEM), and X-ray diffraction (XRD) to evaluate its biomedical properties. The mechanism of the drug pH-responsive release and cytotoxicity were also studied.

The structure of doxorubicin hydrochloride.
Results and discussion
Characterization of MMS
A typical scanning electron microscopy (SEM) image of the Mn-doped mesoporous silica nanospheres is shown in Figure 2. The image shows that the Mn-doped mesoporous silica possesses a uniform spherical shape, with a uniformly distributed particle size and good dispersion.

SEM image of Mn-doped mesoporous silica nanospheres (MMS).
To demonstrate successful doping of Mn, MMS sample was characterized by X-ray photoelectron spectroscopy (XPS) to analyze the state of Mn. As shown in Figure 3, the Mn 2p orbital showed three characteristic peaks at 641.4 eV, 645.5 eV, and 653.1 eV. The two peaks at 641.4 eV and 645.5 eV were indexed to the Mn 2p3/2 orbital. 18 In addition, the Mn 2p1/2 orbital was evident at 653.1 eV which is 11.6 eV different from 641.5 eV, which is similar to the spin orbit splitting energy of Mn3O4. 19

XPS of Mn-doped mesoporous silica (MMS).
To further prove the successful Mn doping, SEM and element mapping of MMS were conducted. It can be seen from Figure 4 that MMS contained silicon, oxygen, and manganese and that the Mn component was uniformly distributed within the shell of MMS (Figure 4(a)–(d)), which showed that manganese had been successfully doped into the framework of silica. The corresponding energy dispersive spectroscopy (EDS) spectrum also confirmed that the MMS contained silicon, oxygen, and manganese and that the Mn-doped amount was 17.3% (mass ratio) (1). These results reasonably verify the existence of Mn as a component in the silica frames.

(a) SEM photograph of MMS. EDS elemental mapping of (b) Si, (c) O, and (d) Mn. (e) The EDS spectrum.
Characterization of MMS-DOX-CMCS
The TEM images of MMS before and after doxorubicin loading are shown in Figure 5. The unloaded MMS was a uniform spherical particle with a size of about 160–175 nm. When the MMS was loaded with doxorubicin, the porous nature disappeared and became invisible, as shown in Figure 5(b). The particle size of MMS-DOX-CMCS was between 300 and 320 nm, being larger than that of MMS. The loading of doxorubicin and encapsulation of carboxymethyl chitosan resulted in MMS having a larger particle size. It is clear that compared to bare MMS, MMS-DOX-CMCS had been coated with a layer of CMCS, showing an obvious core-shell structure.

Transmission electron microscopy (TEM) images of (a) MMS and (b) MMS-DOX-CMCS.
Typical X-ray diffraction (XRD) patterns of MMS, MMS-DOX, and MMS-DOX-CMCS are shown in Figure 6. The major peak 22.5° could be indexed to amorphous silica. 20 The diffraction peak at 33° could be readily assigned to the braunite-1Q (Mn2+Mn3+6SiO12, JCPDS no. 33-0904) phase, 21 which demonstrated that the manganese was successfully incorporated into silica. It was worth noting that after MMS was loaded with doxorubicin and coated with carboxymethyl chitosan, the position of the peak showed no obvious change, indicating that the structure of Mn-doped silica was stable.

XRD patterns of (a) MMS, (b) MMS-DOX, and (c) MMS-DOX-CMCS.
The zeta potential is important for assessing the stability of MMS in suspensions due to the electrostatic repulsion between MMS nanoparticles. 22 As shown in Figure 7, the zeta potential of MMS was about –20.05 mV, which was due to the existence of a large number of silicon hydroxy groups on the surface of silica nanospheres. 23 After being loaded with DOX, the zeta potential of the samples rose to +10.20 mV, which could be attributed to the positively charged amino group of DOX neutralizing the silanol of the silica. At the same time, it also proved the successful loading of DOX. After coating with CMCS, the zeta potential decreased to –21.75 mV due to deprotonation of the carboxylic acid group in CMCS on the silica surface under neutral conditions. Therefore, the charge on the drug loading system was affected by nano silica, doxorubicin, and carboxymethyl chitosan.

Zeta potentials of MMS, MMS-DOX, and MMS-DOX-CMCS.
The thermal properties of MMS, MMS-DOX, and MMS-DOX-CMCS were evaluated by thermogravimetric analysis. The changes in the three samples on heating at a temperature range from room temperature to 800 °C (10 °C min−1) in air were examined. As shown in Figure 8, they all showed a degree of continuous weightlessness. The weight loss occurred in a broad temperature range from room temperature to about 800 °C due to the release of water originating from the particle surface and the intersphere space of MMS. The total weight loss was about 13.81% for MMS, while the total weight loss was about 17.91% for MMS-DOX. The total weight loss of MMS-DOX-CMCS was 21.87%. The high mass loss of MMS-DOX-CMCS was due to the loss of doxorubicin and CMCS during the heating process.
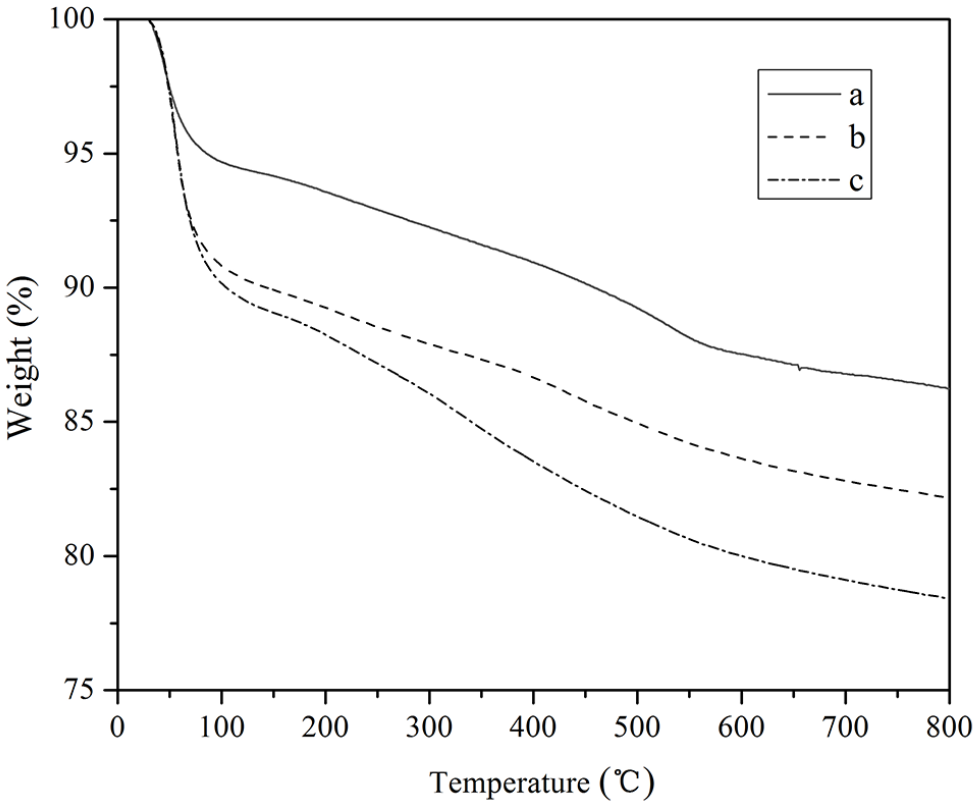
Thermogravimetric weight loss curves of (a) MMS, (b) MMS-DOX, and (c) MMS-DOX-CMCS.
The N2 adsorption method was employed to identify the specific surface areas, pore volumes, and pore diameters of the samples (Figure 9). The N2 adsorption-desorption isotherms of MMS before and after doxorubicin loading are also presented. Both MMS and MMS-DOX-CMCS yielded a type IV isotherm with a H2 hysteresis loop. MMS had a specific surface area of 922.21 m2 g−1 and a pore volume of 0.257 cm3 g−1, which showed that MMS was suitable as a drug carrier. Compared with MMS, MMS-DOX-CMCS had a specific surface area of 197.15 m2 g−1 and a pore volume of 0.170 cm3 g−1. This decrease in surface area and pore volume indicated that DOX had been successfully loaded into the pores of MMS.

N2 adsorption isotherms of (a) MMS and (b) MMS-DOX-CMCS.
As shown in Figure 10, MMS had a narrow pore size of 3–5 nm. This narrow peak shape indicated that the pore size of MMS was relatively uniform. After loading doxorubicin, the pore size distribution did not change significantly.

Pore-size distribution curves of (a) MMS and (b) MMS-DOX-CMCS.
In vitro release of MMS-DOX-CMCS
The high specific surface area and large pore structure of MMS facilitated loading of doxorubicin molecules, and the encapsulation efficiency was 92.73% and the amount of loaded DOX was as high as 18.82%. The release of doxorubicin from MMS-DOX-CMCS was investigated using acidic pH stimulators. Figure 11 shows the cumulative doxorubicin release from MMS-DOX-CMCS in phosphate-buffered saline (PBS) solution under different pH conditions. Over a release time of 48 h, the cumulative release percent of doxorubicin from MMS-DOX-CMCS was 64.53%, 96.50%, and 80.00%, respectively, in PBS buffer solutions with different pH values (5.5, 6.5, 7.4). The cumulative release of doxorubicin from MMS-DOX-CMCS was the result of the joint interaction of outer carboxymethyl chitosan and inner mesoporous SiO2. 24 When the hydrogen ion (H+) concentration was high (pH = 5.5), the presence of H+ led to curling of the molecular chain of carboxymethyl chitosan, which resulted in the swelling capacity decreasing, and the release of DOX was blocked. The release was mainly the outward diffusion of DOX in mesoporous MMS, and therefore, the overall release was low. With an increase in pH, the swelling rate of carboxymethyl chitosan increased, and the cumulative release rate of doxorubicin from MMS-DOX-CMCS gradually increased. When the pH was 6.5, the release amount of DOX was the largest. Moreover, the acidic environment was conducive to the dissolution of manganese as well as cleavage of the Mn–O bond of MMS-DOX-CMCS, which led to degradation of the Si–O–Si framework. The extracellular environment of most tumors is weakly acidic (pH value is about 6.5); therefore, MMS-DOX-CMCS has a good application prospects. When the pH was 7.4, the release decreased. The possible reasons are as follows: the carboxyl groups in carboxymethyl chitosan were deprotonated and dissociated into carboxyl ions (–COO–), and the electrostatic repulsion between –COO− increased the particle size of MMS-DOX-CMCS.25,26 The thick outer layer of MMS-DOX-CMCS lengthened the diffusion path and hindered the release of doxorubicin from the mesoporous SiO2. 27 The above results show that the release of doxorubicin from MMS-DOX-CMCS had an obvious pH response and that the drug release could be controlled by changing the pH value.

In vitro release profiles of MMS-DOX-CMCS at different pH values.
For MMS-DOX, the release rate of doxorubicin was faster compared with MMS-DOX-CMCS, as shown in Figure 12, which indicated that carboxymethyl chitosan had the effect of delaying drug release.

In vitro release profiles of MMS-DOX at different pH values.
Cytotoxicity
A typical 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) method was used to assess the in vitro cytotoxicity of MMS. As shown in Figure 13, the cell viability of HepG2 cells was still maintained at 92.9%, even after co-incubation with MMS at a high concentration of 200 μg mL−1, indicating that blank MMS was sparingly toxic.
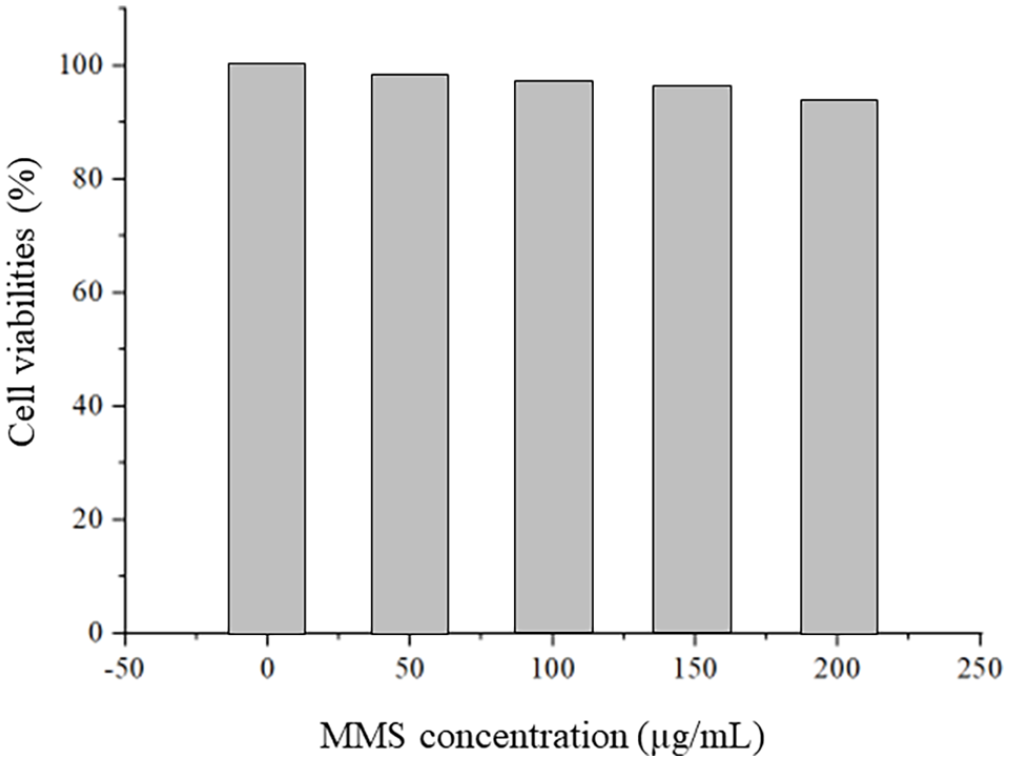
Cell viabilities of Hep G2 cells after co-incubation with MMS.
Compared to the equivalent concentrations of free DOX, the cells co-incubated with DOX-loaded MMS-DOX-CMCS showed much lower viabilities (Figure 14), indicating that the MMS-DOX-CMCS drug delivery system could result in a substantially enhanced therapeutic efficacy of DOX against Hep G2 cancer cells.

Cytotoxicity of MMS-DOX-CMCS and free DOX against Hep G2 cells.
Conclusion
In summary, pH/responsive, biodegradable, Mn-doped mesoporous silica (MMS) nanospheres have been prepared via an in situ doping route. The results of TEM and N2 adsorption revealed that empty MMS possessed a spherical shape, a highly porous structure, a large specific surface area of 922.21 m2 g−1, and a pore volume of 0.257 cm3 g−1. The unique features of MMS led to them having high loading efficiency (18.82%) for doxorubicin. MMS-DOX-CMCS can respond to the acidic environment of tumor tissues and realize simultaneously on-demand drug release resulting from the rapid cleavage of Mn–O bonds and the good swelling properties of the carboxymethyl chitosan coating layer. This work not only provides a new method for drug delivery but has also improved the development of drug delivery systems.
Experimental details
Materials
All solvents and reagents, unless otherwise stated, were of analytical grade and used without further purification. Cetyltrimethylammonium bromide (CTAB) and ethanol were purchased from Sinopharm Group Chemical Preparation Co., Ltd. Triethanolamine (TEA), tetraethyl orthosilicate (TEOS), 3-aminopropyltriethoxysilane (APTES), dimethylsulfoxide (DMSO), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), doxorubicin (DOX), manganese sulfate (MnSO4), and carboxymethyl chitosan were obtained from Shanghai Maclean Biochemical Technology Co., Ltd. Distilled water was used for all experiments.
Synthesis of Mn-doped mesoporous silica (MMS)
The Mn-doped mesoporous silica was prepared according to the literature. 15 In a typical synthetic procedure, 2.182 g of CTAB and 1.1 mL of TEA were dissolved in a mixed solution composed of 2 mL of ethanol and 20 mL of H2O. Under stirring conditions, 10 mL of MnSO4 solution (8 mg mL−1) was then added dropwise to the above mixture. Subsequently, the mixture was heated to 80 °C and 1.5 mL of TEOS was slowly added dropwise. After reacting for 4 h, the brown product was collected by centrifugation (9000 rpm, 10 min) and washed several times with water and ethanol to remove the residual reactants. Finally, the sample was dried under vacuum at 40 °C for 24 h. The dried sample was calcined at 550 °C for 6 h in a muffle furnace to remove the template and give the desired Mn-doped mesoporous silica nanospheres.
Preparation of MMS-NH2
0.2 g of MMS was dissolved in 20 mL of ethanol to form a solution which was ultrasonicated at 500 W for 30 min. Subsequently, the mixture was heated at 80 °C and 0.18 mL of APTES was slowly added dropwise. After refluxing for 24 h, the product was collected by centrifugation (9000 rpm, 10 min), washed several times with water and ethanol, and dried under vacuum at 40 °C for 24 h.
Drug loading
The loading of DOX was accomplished by diffusion and electrostatic adsorption. 30 mg of MMS-NH2 and 20 mg of DOX were dispersed into 10 mL of distilled water to form a suspension that was ultrasonicated for 20 min and then stirred for 24 h at room temperature in the dark. The resulting product (MMS-DOX) was collected by centrifugation (9000 rpm, 10 min) and washed three times with water and ethanol to remove the excess DOX. Finally, the DOX-loaded MMS-NH2 was dried under vacuum for 24 h and named MMS-DOX. The supernatant and all washing solutions were collected, and the remaining amount of DOX was measured with a UV-Vis spectrophotometer at 481 nm to calculate the drug loading efficiency.
Carboxyl activation of carboxymethyl chitosan
10 mg of carboxymethyl chitosan was added to 10 mL of ultrapure water. 25 mg of EDC and 15 mg of NHS were added to the above mixture which was then stirred for 6 h to activate the carboxyl group.
Encapsulating carboxymethyl chitosan on MMS-DOX
MMS-DOX was mixed with activated carboxymethyl chitosan to form a suspension which was shaken for 24 h at room temperature. The resulting product (MMS-DOX-CMCS) was collected by centrifugation (9000 rpm, 10 min) followed by drying under vacuum for 24 h.
In vitro drug release
The in vitro release of MMS-DOX-CMCS or MMS-DOX was investigated using the dialysis method. 28 The drug-loading and drug-entrapment ratios were also determined. 8 mg of MMS-DOX-CMCS were added to 10 mL of phosphate buffer solution (PBS). The resulting mixture was sealed in a dialysis bag (M > 8000 Da) and the bag was fixed on paddles and placed in a 50 mL buffer solution of pH 5.5. Following incubation was carried out at 25 °C; 3 mL samples were withdrawn at predetermined time intervals from each vessel and the same volume of fresh PBS was added to the vessel to replace the lost fluid. The drug-loading and drug-entrapment ratios were determined by UV-Vis illumination (UV-2401PC, SHIMADZU, Japan) at a wavelength of 481 nm. A blank control was used as a reference. The procedure was the same as that when employing a sample at pH 5.5, but the pH was 6.5 and 7.4, respectively.
The preparation, release, and degradation of the MMS-DOX-CMCS system is shown in Scheme 1.

The preparation, release, and degradation of the MMS-DOX-CMCS system.
The drug-loading ratio and drug-entrapment ratios were calculated using the following formulae:

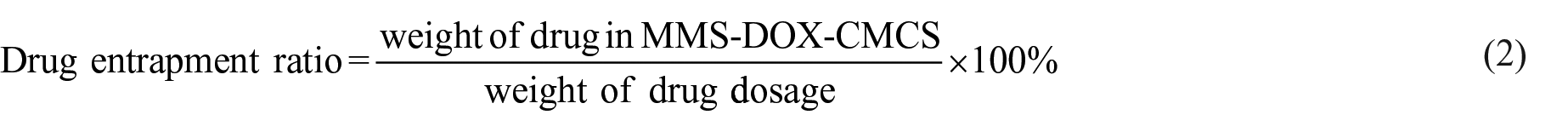
Characterization methods
UV-Vis absorption spectra were obtained using a UNICO WFZ UV-2802PC/PCS spectrometer. Powder X-ray diffraction (XRD) measurements were performed on a Rigaku D/MAX-PC 2500 diffractometer with Cu Kα radiation (λ = 0.15405 nm) from 10° to 80° at a scanning rate of 5°/min. Transmission electron microscopy (TEM) micrographs were obtained using a JEM-2100F electron microscope operating at 200 kV. X-ray photoelectron spectroscopy (XPS) measurements of the Mn-doped mesoporous silica nanospheres (MMS) were recorded by using a Kratos XSAM-800 spectrometer with Mg Kα radiation. The crystallization behavior was monitored using a PerkinElmer thermogravimetric (TG) instrument, scanning in the temperature range from room temperature to 800 °C at a rate of 10 °C per minute. Field emission scanning electron microscopy (FE-SEM) and EDS were carried out using a field emission microscope (JEOL, 7500B) operating at an acceleration voltage of 10 kV. The specific surface areas, pore volumes, and pore diameters of the samples were determined according to the Brunauer–Emmett–Teller and Barrett–Joyner–Halenda procedures employing the N2 adsorption method using a high-speed automated surface area and pore size analyzer (NOVA 2000, Quantachrome, USA). Zeta potential values were obtained with a Zeta sizer Nano ZS (Malvern, UK) at 25 °C.
Cell culture studies
HepG2 cells were cultured in RPMI-1640 medium complemented with 10% fetal bovine serum (FBS). Cells were incubated at 37 °C under an atmosphere comprising 5% carbon dioxide (CO2). 29 The medium was replaced every 24 h before the cells were passaged.
Cytotoxicity assay
The cytotoxicity was evaluated by the MTT assay. HepG2 cells were seeded in a 96-well plate at a density of 10,000 cells per well and incubated for 24 h at 37 °C with 5% CO2. Next, MMS-DOX-CMCS and free DOX were added to the cells in RPMI-1640 medium supplemented with 10% FBS at different concentrations, respectively, and incubated at 37 °C with 5% CO2 for 24 h. Untreated cells in growth media were used as the blank control. Subsequently, MTT solution was added to each well and the 96-well plate was incubated for another 4 h at 37 °C. The RPMI-1640 medium was removed and 100 mL of dimethylsulfoxide (DMSO) was added and the wells shocked for 10 min. The optical density (OD) of each well was measured at 570 nm by using a TECAN Infinite F200 Microplate Reader. 30
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Education Department of Jilin Province (JJKH20220879KJ and JJKH20210977KJ). This work was supported by Innovation and Entrepreneurship Training Program for College Students in 2022 (202210199030) and in 2021 (S202110199086). This work was supported by the Traditional Chinese Medicine Science and Technology Project of Jilin Province (Grant Nos 2022006 and 2021007).