
Abstract
Proline is an important amino acid that plays unique roles in the structures of peptides and proteins. The conformations of proline are searched by a thorough method, generating 3888 trial structures optimized at the B97D/6-311++G** level. A total of 23 conformations are found and their structural and energetic data are presented. All the proline conformers exhibit a coplanar feature for four of the five pyrrolidine ring atoms. The coplanar rule reduces the cost of the conformational search by a factor of 40. The theoretical composition-weighted infrared spectrum provides a good explanation of the experimental results. A conformational search of capped proline yields seven unique conformers, all with trans C-termini peptide planes. The trans C-termini rule further cuts by half the cost of the conformational search of proline-containing peptides. The theoretical composition of the cis N-termini peptide bonds at room temperature is 5.5%, agreeing with the experimental estimations of 3%–10%.

Introduction
Among the 20 proteogenic natural amino acids (AAs), proline (P) is unique in forming the secondary structure of peptides and proteins due to its important pyrrolidine ring. In addition to being required for protein biosynthesis, proline plays critical roles in cellular bioenergetics,1–5 osmoregulation,5,6 stress protection,7–10 and cellular signaling processes.3,11,12 Studies over the last two decades have also established the specialized role of proline in cancer metabolism.3,12–16 Recent discoveries of the broad effects of proline metabolism on cancer cell growth and survival have implicated proline metabolic enzymes as potential targets for therapeutic intervention.17–21
Proline is also involved in many antimicrobial peptides (AMPs). AMPs are small peptides with a broad spectrum of antibiotic activities against bacteria, fungi, protozoa, and viruses, and sometimes exhibit cytotoxic activity toward cancer cells. AMPs containing predominantly either tryptophan or proline can kill microorganisms by targeting intracellular pathways and are, therefore, a promising source of next-generation antibiotics. Proline-rich AMPs interact with the 70S ribosome and disrupt protein synthesis. They can also target the heat shock protein in target pathogens, and consequently lead to protein misfolding. 22
The special biological functions of proline are intimately related to the conformations it may adopt. Because of the existence of a pyrrolidine in its ring side chain, proline is less flexible than other AAs. Comparing with other AAs, an amidogen H atom is replaced by a C atom in proline, and it is difficult for proline to form a hydrogen bond (H-bond) with the C–O of the fourth residue at the C-terminal or the nitrogen on an adjacent peptide chain. As a result, proline tends to destroy the α-helix or the β-fold and ends up at the end of a protein secondary structure.
Besides, due to the absence of the N–H bond and the associated spatial repulsion of amide H, the cis conformation may appear in a peptide containing proline, while it is basically forbidden in other peptide bonds. The cis populations of proline in water and chloroform have been established by the experiments of Delaney and Madison. 23 They found about 25% cis peptide bond isomers in water and 15%–20% cis in chloroform. Park and Kang 24 found that their density functional theory (DFT) results were consistent with experiments. For gas phase proline, the fraction of cis X–P peptide bonds is typically in the range of 3%–10%25,26 (X stands for any AA), and may be up to 40% for some aromatic AA–P peptide bonds. 27
Considering the fundamental importance of proline as an elemental AA and its unique features in peptide and protein structures, a detailed knowledge about the conformations of natural and capped proline is desirable. Capped proline refers to a proline capped by a peptide bond at both its N- and C-termini and mimics proline in a peptide or protein better than in natural proline. As illustrated by a number of studies,28–30 the structural properties of natural and capped proline are substantially different. A conformational study of capped proline is essential for understanding the behavior of proline in peptides and proteins. As experimental approaches such as infrared (IR) spectroscopy, mass spectrometry ultraviolet (UV) spectroscopy, and electric dipole moment measurements generally provide only indirect structural information, computational explorations of proline conformations are of special importance.
Sapse et al. 31 carried out the earliest ab initio study of proline conformations at the RHF/STO-3G and RHF/6-31G levels. Ramek et al. 32 conducted a search on the potential energy surface (PES) at the RHF/6-311++G** level and found 10 proline conformers. They also determined the reaction paths and potential energy barriers connecting the PES stationary points. Császár and Perczel 33 obtained 12 proline conformers and their relative energies at correlated levels of electronic structure theory. Meanwhile, Stepanian et al. 34 identified 15 stable minima on the PES of proline and reassigned the vibrational spectra of unionized proline obtained from matrix-isolation IR spectroscopy. Lesarri et al. 35 configured a special laser-ablation system to obtain the rotational parameters of two plausible gas phase proline conformers and calculated their structures by an iterative-fit procedure. Czinki and Császár 36 found 18 proline conformers at the DFT(B3LYP)/6-311++G** level of theory. The search of proline conformations by Czinki and Császár is the most thorough so far. However, their search did not consider the variations of the dihedral angles (DAs) for the main chain Cα–C and C–OH bonds, and some energy minima may be easily missed in their search. Recently, Ten and Shcherbakova 37 performed B3LYP/6-311++G (d, p) calculations of the IR spectra of three proline conformers and attempted to attribute the experimental spectrum to two of the three proline conformers. However, the three conformers do not include the global minimum of the conformation ensemble of Czinki and Császár. 36 Not surprisingly, the comparison between the computational result of Ten and Shcherbakova 37 and the experimental spectrum is of low quality.
Computational studies of the conformations of capped proline are less common than that of natural proline. Yuan et al. 38 provided a comprehensive conformational analysis of the 20 capped AAs. In this outstanding work, they identified five unique conformations for capped proline. Unfortunately, no connection was made between the computed conformations and the unique structural characteristics of proline in peptides.
In this study, the PESs of natural and capped prolines are thoroughly explored and the structures of all energy minima are analyzed. The equilibrium distributions of the conformational ensemble at various temperatures are presented. In addition, the theoretical and experimental IR spectra of natural proline are compared. The ensemble of computed conformations of capped proline is used to explain quantitatively the compositions of the cis–trans structures of proline in peptides.
Computational methods
To examine the characteristics of proline conformers comprehensively, the conformational space of natural proline is explored through a systematic variation of all its seven bond rotational degrees of freedom, as indicated in Figure 1(a). Details about the bond rotations and the values of the DAs used are summarized in Table 1. By considering all possible combinations of the DAs, a total number of 4 × 2 × 3 × 3 × 3 × 3 × 3 × 2 = 3888 trial structures are generated and used for exploring the PES of proline. The trial structures are optimized at the B97D/6-311++G** level of theory, with the default convergence criteria of Gaussian09, for example, maximum force <0.00045, root mean square (RMS) of force <0.0003, maximum displacement <0.0018, RMS of displacement <0.0012. The B97D/6-311++G** level of theory is chosen as it has been shown to provide the required computational accuracy for AAs and peptides.39,40 The set of unique conformations thus obtained are verified to be true energy minima by frequency calculations as well as further geometry optimizations at the B97D/6-311++G** level. Both the electronic energies and the free energy corrections at the B97D/6-311++G** level are used to determine the equilibrium conformation distributions at various temperatures. 41 The harmonic approximation of molecular vibrations is used, that is, the conformer structures and their vibrational modes are temperature independent.

Planar structure and atom numbering for: (a) proline (P), with all the rotational degrees of freedom indicated (see Table 1 for more details); (b) capped P, with the bond rotations necessary for the conformational search indicated (see the main text for more details).
Bond rotations and the dihedral angles used in the generation of the trial structures for the conformational search of proline.

The vibrational frequencies of the proline conformations are calculated at the B97D/6-311++G** level. Based on the scaling factors suggested for B3LYP/aug-cc-pvdz 37 and through benchmarking tests, the B97D/6-311++G** frequencies are scaled with a scaling factor of 0.97 for the OH, NH, and CH stretching modes, and with a scaling factor of 1.01 for all other vibrations. The infrared spectrum for the conformational ensemble is calculated from the equilibrium compositions and smoothed with a 5-point Savitzky–Golay smoothing function. 42
The conformational search of capped proline is conducted in a way much more efficient than that for proline. Based on analysis and testing of the obtained proline conformers, only two of the five pyrrolidine ring bond rotations are necessary, as indicated in Figure 1(b). Besides, the bond rotations b(C3–C4–O5–H) and h(H–N8–C3–C4) in proline are now revised, respectively, to b(C3–C4–N5–C12) and t(C11–C9–N8–C3) due to the changed C- and N-termini in the capped proline. The number of trial structures required for the PES exploration of capped proline is thus reduced to 96, and can be further reduced to 48, as discussed in the “Results and discussion” section. The trial structures of capped proline are also optimized at the B97D/6-311++G** level.
All the trial structures used in this study are generated with our in-house developed software written in Python consisting of 11 subroutines and 1576 lines of computing code. All the DFT calculations are carried out using the Gaussian-09 software package. 43
Results and discussion
Conformations of proline
A total of 23 conformers are found through the PES exploration. All the conformers are unique as assessed from their energy–dipole moment combinations and verified by final visual inspection. The relative electronic energies (ΔE), zero-point vibrational energies (ΔVE0), Gibbs free energies (ΔG0) at room temperature (T0), rotational constants and permanent dipole moments of all 23 proline conformers are shown in Table 2. As seen in Table 2, all the 23 conformers fall within an energy range of 10 kcal mol−1. For the relatively important low-energy conformers that are less than 3 kcal mol−1 above the global minimum, there are five conformers if measured by ΔE, or seven conformers if measured by ΔG0.
Relative electronic energies (ΔE, kcal mol−1), relative ZVPEs (ΔVE0, kcal mol−1), relative Gibbs free energies at room temperature (ΔG0, kcal mol−1), rotational constants (GHz) and permanent dipole moments (D, Debye) of the proline conformers determined at the B97D/6-311++G** level of theory.

The 23 conformers here include all the 18 conformers reported by Czinki and Császár. 36 Conformers 5, 7, 19, 20 and 22 are newly identified in this search. Among the five lowest-energy conformers, the newly identified conformer 5 has the most favorable free energy correction (Δg), as may be inferred by comparing Δg0 (Δg at T0) = ΔG0 – [ΔE + ΔVE0] in Table 2. Specifically, Δg0 is −1.0, −0.71, −0.9, and −0.26 kcal mol−1 for conformers 5, 4, 3, and 2, respectively. As Δg ~ −Δ(TS), conformer 5 is expected to be increasingly favorable with an increase in the temperature. In fact, as can be seen in Table 3, with an equilibrium population of over 10%, conformer 5 is important for interpreting experimental data measured at T > 400 K. 44
Equilibrium compositions (%) of proline conformers.

The structures of the four lowest-energy conformers and the five newly found conformers, together with their H-bond lengths, are shown in Figure 2. Instead of using popular but complicated H-bond rules,45,46 the H-bond here is assumed to exist if the distance between the H-bond donor and acceptor is below the cutoff criterion of 2.8 Å. The distance-only criterion is also widely used and is adopted here as it is not only simple, but also capable of explaining the energy difference between cis- and trans-carboxy conformations. 47 The rule about the H-bond angle of more than 110° 48 is ignored here as it disagrees with the experimental observations on H2O. . .HF and H2S. . .HF 49 and is optional according to the International Union of Pure and Applied Chemistry (IUPAC) Recommendations 2011. 50
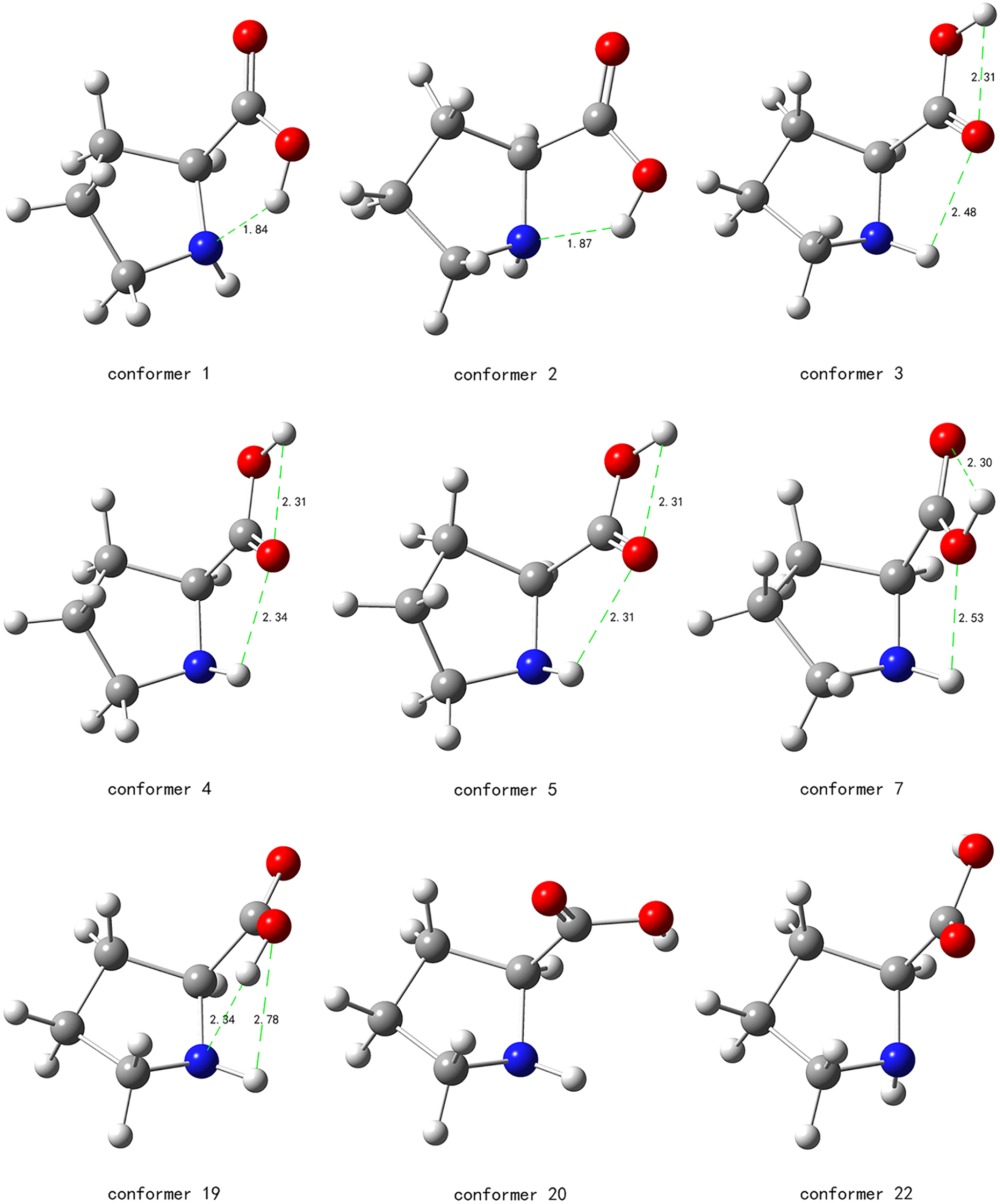
Representative conformations of proline (P). Conformers 5, 7, 19, 20, and 22 are newly found in this work. H-bonds are indicated by dashed green lines and the H-bond lengths are given in Å.
As seen in Figure 2, both conformer 1 and conformer 2 have O–H. . .N H-bonds that are quite short. Conformers 3, 4, and 5 all possess the O–H. . .O6 and N–H. . .O6 H-bonds. The O–H. . .O6 and N–H. . .O6 H-bond lengths are similar but are about 0.5 Å longer than the O–H. . .N H-bond. Apparently, the O–H. . .N H-bond is much stronger than both the O–H. . .O6 and N–H. . .O6 H-bonds. The strong O–H. . .N H-bond is the main reason for the low energies of conformers 1 and 2.
The equilibrium distributions of the proline conformers at temperatures of 98, 198, 298, 398, 498, and 548 K are shown in Table 3. The conformer distribution at 548 K (275°C) is needed when comparing the theoretical and experimental IR spectra. As shown in Table 3, proline exists predominantly as conformer 1 and conformer 2 at T ⩽ T0. The compositions of conformers 3, 4, and 5 increase notably with T. Their combined compositions are 2%, 13%, 28%, 38%, and 40% for T = 198, 298, 398, 498, and 548 K, respectively.
The pyrrolidine ring structure and an efficient method of searching proline conformations
Examining the structures of all proline conformers, as partly shown in Figure 2, a universal feature is observed. That is, four of the five pyrrolidine ring atoms are very close to existing in a common plane. Denoting the five pyrrolidine ring atoms as A, B, C, D, and E, where A, B, C, and D lay approximately in a plane and E is the non-coplanar atom. The deviation of the four atoms from forming an ideal plane may be characterized with γ = abs(DA(A–B–C–D)), referred to below as the non-planeness. The procedure of finding A, B, C, D, E, and γ is straightforward. The five DAs concerning the five pyrrolidine ring atoms, DA(C1–C2–C3–N8), DA(C2–C3–N8–C7), DA(C3–N8–C7–C1), DA(N8–C7–C1–C2), and DA(C7–C1–C2–C3), were computed and compared. The four atoms yielding the smallest absolute DA value are said to be coplanar atoms, and the corresponding DA is referred to as the non-planeness, γ. For example, γ = abs(DA(C2–C3–N8–C7)) if it is the smallest among the five DAs. The four atoms, C2, C3, N8, C7, are coplanar, and are referred to as A, B, C, D, respectively. The other pyrrolidine ring atom, C1, is the non-coplanar atom, E.
The non-planeness and the distances (d) between E and the mean ABCD plane are shown in Table 4 for all 23 proline conformations. Here d is calculated as the average of the distance between E and the plane (ABC) and the distance between E and the plane (BCD). The sign of d is positive if E and C4 are on the same side of the mean ABCD plane, and negative otherwise.
Pyrrolidine ring structures of proline conformers: the non-coplanar atom (E), the non-planeness (γ, °) and the distance between E and the ABCD plane (d, Å).

See the main text for definitions.
As seen in Table 4, the non-planeness is generally quite small, with a maximum of only 11.4° for conformer 8 and an average of 6° for all 23 conformers. The coplanar rule is, therefore, a fairly good approximation. In addition, it is noted that C2 is always one of the coplanar atoms, as shown in Table 4. Besides, the d values are all positive except for those of conformers 2 and 6.
The coplanar feature means that the five bond rotations, (c, d, e, f, g), about the pyrrolidine ring are inter-dependent, and we are left with only two independent bond rotations. Therefore, the eight bond rotations (a, b, c, d, e, f, g, h) shown in Figure 1(a) can be reduced to five independent bond rotations (a, b, c, d, h) in Figure 1(b). The number of full combinations of the rotational degrees of freedom is thus reduced to 4×2×3×3×2=144. The number of trial structures for the conformational search can, in fact, be further reduced. Notice that the bond rotation c (d) corresponds to the swing of C7 (C1) relative to the N8–C2–C3 plane. Specifically, c = −90°, −120°, and −150° indicate that C7 is positioned above, on, and below the N8–C2–C3 plane, respectively. Meanwhile, d = 90°, 120°, and 150° correspond to the fact that C1 is positioned above, on, and below the N8–C2–C3 plane, respectively. The four-atom coplanar rule can be satisfied if either C1 or C7 is on the N8–C2–C3 plane, or when C1 and C7 are both above or both are below the N8–C2–C3 plane. That is, only seven of the nine combinations of (c, d) may satisfy the four-atom coplanar rule. As the (−120°, 120°) combination means a five-atom coplanar form is absent in any proline conformations, only six of the (c, d) combinations are useful for the conformational search. Consequently, Table 1 can be replaced by Table 5 for the search of neutral proline conformations. And the total number of bond rotation combinations is 96 (= 4×2×6×2), a 40-fold reduction from the initial number of 3888. Table 5 has been tested to fully reproduce the conformational search results of Table 1.
Bond rotations and dihedral angles for the conformational searches of proline (P) and capped P.

The pyrrolidine ring remains unchanged with the formation of a peptide bond at the N- and/or C-terminus of P. The four-atom coplanar rule of the pyrrolidine ring is also expected to hold for proline in peptides. The bond rotations for the conformational searches of proline and capped proline are, therefore, basically the same, except that the bond rotation h (H–N8–C3–C4) for proline is changed to t (C11–C9–N8–C3) for capped P, as shown in Figure 1 and Table 5. Consequently, the number of trial structures generated for the conformational search of capped proline is 96, the same as that for natural P.
It should be pointed out that the puckered structures of the proline ring have been well-established in terms of the concept of pseudorotation by DeTar and Luthra. 51 They described the conformational state of the five-membered ring of the proline side chain using only two parameters. However, the new four-atom coplanar rule of the pyrrolidine ring revealed in this study is more advantageous in reducing the size of the conformational search space.
Vibrational spectrum of natural proline
The IR spectra of the four conformers 1, 5, 7, 22 and the weighted average of the 23-conformation ensemble at T = 548 K are shown and compared with the experimental result (Figure 3).

Simulated IR spectra of four representative conformers and the conformational average of proline at 275°C, as well as the experimental results of Linder et al. 44 The theoretical IR bands are Gaussians with a full width at half maximum of 8 cm−1. IR: infrared.
Conformers 1, 5, 7, and 22 have different H-bonds (Figure 2), resulting in different frequencies of the related vibrational modes (Figure 3). Due to the strong O–H. . .N H-bond of conformer 1, its O–H stretching mode, q(OH), shows a large red-shift, as seen in Figure 3. Specifically, the q(OH) frequencies are 3099, 3566, and 3560 cm−1 for conformers 1, 5, and 7, respectively, in comparison with 3597 cm−1 for conformer 22 with no H-bond. That is, the strong O–H. . .N H-bond causes a red-shift of about 500 cm−1 for q(OH) of conformer 1. The O–H. . .N H-bond, however, inhibits the flexibility of the O–H in-plane-bending motion (δ(OH)), causing a blue-shift for δ(OH) of conformer 1. The δ(OH) frequencies are 1419, 1125, 1120, and 1269 cm−1 for conformers 1, 5, 7, and 22, respectively.
Notice that both the blue-shifted O–H bending of conformer 1 and the red-shifted bending of conformer 5 have high IR intensities (Figure 3). They are critically important for the correct interpretation of the experimental results. For example, the vibration for νexp ≈ 1367 cm−1 is missing in the computation of Linder et al. 44 who used only one conformer in their calculations. Similarly, νexp ≈ 1110 cm−1 cannot be properly assigned to δ(OH) in the study by Stepanian et al. 34 and in that by Ten and Shcherbakova. 37
It is of practical interest to emphasize that conformations with similar H-bonds also produce similar IR spectra, as is well-documented in the literatures, 52 and is also seen in Figure 3 for conformers 5 and 7. That is, as far as the IR spectrum is concerned, the ensemble average may be approximated by a mix of a few conformers with a proper coverage of characteristic H-bonds. For example, Table 3 shows that only conformers 1–5 are significant at T = 548 K, while Figure 2 shows that the H-bonds in conformers 1 and 2 or in conformers 3–5 are similar. At T = 548 K, the combined populations are 36.6% for conformers 1 and 2 and 37.7% for conformers 3, 4, and 5. Using a 50%/50% mix of the IR spectra of conformer 1 (or 2) and conformer 3 (or either 4 or 5) produces roughly the same IR spectrum as the ensemble average shown in Figure 3.
Table 6 compares the experimental and theoretical frequencies for the major IR peaks observed, together with their vibrational mode assignments. Notice that there are several C–H bonds, each with a somewhat distinct stretching frequency, forming a broad q(CН) band. The lowest and highest C–H stretching frequencies are denoted in Table 6 as q(CH)L and q(OH)H, respectively.
Characteristic IR modes of proline at 548 K: experiment (νexp, cm−1) versus theory (νcal, cm−1). q(CH)L and q(OH)H denote, respectively, the lowest and highest C–H stretching frequencies.

IR: infrared.
As seen in Table 6, the experimental and theoretical results are in excellent agreement. Very good agreement between the experimental and theoretical IR spectra is also seen in Figure 3, except that the theoretical q(OH) peaks of conformers 1 and 2 are absent in the experiment. The complete absence of the theoretical q(OH) = 3099/3157 cm−1 in the experimental IR spectrum is quite puzzling when considering the overall high quality theoretical and experimental agreement shown in Figure 3. One possible reason is that the scaling factor for q(OH) associated with a strong H-bond should be smaller than the value of 0.97 assumed above. As noted by Stepanian et al., 34 theoretical and experimental comparisons of q(OH) affected by a strong H-bond yields a scaling factor of 0.92 for glycine, alanine, and valine, instead of the value of 0.96 suggested by calculations at the B3LYP/aug-cc-pVZD level.49,53,54 Benchmarked with 0.92 instead of 0.96 for B3LYP/aug-cc-pVZD, the scaling factor here for q(OH) of conformer 1 or 2 should be reduced to 0.93, instead of 0.97 as used above. With the newly adjusted scaling factor, q(OH) is found to be 2971/3027 cm−1 for conformer 1 or 2. The revised q(OH) value is close to q(CH)H = 2967 cm−1 and would merge with the q(CH) modes to form the experimentally observed broad band of νexp ~ (2800 cm−1, 3000 cm−1) shown in Figure 3. The theoretical and experimental IR spectra would, therefore, be in a perfect agreement.
Conformations of capped proline
Seven conformers are found for the capped proline, adding only the relatively unimportant conformers 5 and 7 to the existing results of Yuan et al. 38 To facilitate further discussion, the structures and the intra molecular H-bonds of all the seven conformers of capped proline are shown in Figure 4.

Conformations of capped proline (P). Intramolecular H-bonds are noted with dashed green lines. The H-bond lengths are also indicated.
As shown in Figure 4, the C-termini peptide plane of capped proline, O6C4N5H, always adopts the trans form. Moreover, the capped C-termini can only serve as H-bond donor, but not as an acceptor. The trans form and H-bond donor restrictions are responsible for the small number of conformations of capped proline. As with natural proline, four of the five pyrrolidine ring atoms of capped proline are coplanar, with a maximum deviation of 11.8° for conformer 2 and an average deviation of 6.6° for all the conformations. Due to the influence of the N-terminus capping group, however, both N8 and C3, instead of C2 for natural proline, are always the coplanar atoms. Besides, C4 and the non-coplanar atom, C1 or C2, are on the opposite side of the four-atom plane for six conformers of capped proline. Only for conformer 2 with C7 as the non-planar atom are C4 and C7 on the same side of the four-atom plane.
As the C-termini peptide plane of all conformations of capped proline unanimously adopt the trans form, the bond rotation b is unnecessary for the conformational search of capped proline. As a result, the number of trial structures required is reduced from 96 to 48. The 48 initial trial structures are verified to reproduce all the seven conformations of capped proline. The reduction in the number of trial structures is quite important for the conformational searches of proline-containing peptides.
The relative electronic energies, zero-point vibrational energies, Gibbs free energies at T0, rotational constants and permanent dipole moments of all the capped proline conformers are shown in Table 7. As seen in Table 7, the seven conformers fall within an energy range of 6 kcal mol−1 when measured by ΔG0.
Relative electronic energies (ΔE, kcal mol−1), relative ZVPEs (ΔVE0, kcal mol−1), relative Gibbs free energies at T0 (ΔG0, kcal mol−1), rotation constants (GHz) and permanent dipole moments (D, Debye) of the capped proline conformers determined at the B97D/6-311++G** level of theory.

The equilibrium distributions of capped proline conformers at temperatures between 98 and 548 K are shown in Table 8. As shown in Table 8, the capped proline exists predominantly as conformer 1 at T ⩽ T0. The compositions of conformers 2, 3, and 4 increase notably with T. Their combined compositions are 2%, 11%, 24%, 35%, and 42% for T = 198, 298, 398, 498, and 548 K, respectively.
Equilibrium contents (%) of the conformers of capped proline.
Cis and trans peptide bond structures of proline-containing peptides
As shown in Figure 4, the C-terminus peptide plane adopts the trans form for all capped proline conformations, but the N-terminus peptide plane may be in either the trans or the cis form. Specifically, the N-termini peptide bonds of conformers 1, 2, and 5 are trans, while those of conformers 3, 4, 6, and 7 are cis. Notice that the N- and C-termini peptide planes are, respectively, the analogue of the X–P and P–Xnp peptide planes in a general peptide chain. Here, X–P (P–Xnp) means X (Xnp) is connected to P and on the N (C)-terminal side of P. X can be any AA residue, while Xnp can also be any AA residue excluding P. The trans–cis contents of the capped proline conformations can be used to mimic the populations of trans–cis X–P and P–Xnp peptide planes.
Using the equilibrium distributions shown in Table 8, the proportions of the cis X–P and P–Xnp peptide bonds at different temperatures can be calculated and are displayed as the blue and orange lines in Figure 5, respectively. As shown in Figure 5, the population of cis X–P peptide bonds increases with the temperature, and is 5.5% at T0. The computational results agree well with the early observation of Chen and Bovey that there are 5%–10% cis structures for the X–P peptide plane, but essentially nil for the P–Xnp peptide plane. 26 The theoretical results are also consistent with the recent report by Alderson et al. 25 By using multi-dimensional nuclear magnetic resonance (NMR) to quantify the cis–trans peptide bonds in unfolded proteins at T0, Alderson et al. found that peptide bonds are always trans, except for 3%–10% cis structures for the X–P peptide bonds.

Percentages of the equilibrium contents of X–P and P–Xnp conformations with cis peptide bonds.
The experiment of Jabs et al. 55 estimated that there are about 0.03% cis peptide bonds for P–Xnp. Although their results were not exactly the same as our result of 0% cis P–Xnp, they do agree quite well. The minor theoretical and experimental discrepancy indicates that the environment experienced by the P–Xnp peptide bond in a long peptide chain may sometimes induce a potential energy barrier for the bond rotation b. Nevertheless, the experiment does show clearly the energy of the cis configuration, when as a local minimum of the PES, is much higher than that of the trans form. The cis form of the P–Xnp peptide plane can almost always be neglected. A total of 48 trial structures per proline residue suffice for the full conformation search of proline-containing peptides, such as proline-containing AMPs.
Due to a lack of definite and precise measurements, a detailed quantitative theoretical, and experimental comparison cannot be made yet. However, the computational results here do provide, for the first time to our knowledge, a very good quantitative explanation of the experimentally observed population of cis X–P peptide bonds.
Conclusion
Through comprehensive PES exploration, a total of 23 proline conformations have been found, among which 5 conformers are newly identified. The 23 conformations span an energy range of 10 kcal mol−1, with 5 of them existing less than 3 kcal mol−1 above the global minimum. The relative energies, rotation constants, dipole moments, relative ZPVEs, relative Gibbs free energies,and equilibrium distributions of the proline conformations as determined at the B97D/6-311++G** level are presented. Structural analysis reveals a coplanar rule for four of the five atoms of the pyrrolidine ring. The coplanar rule can reduce the computational cost of the conformational search by a factor of 40, while producing the same results as those of the thorough search method. The equilibrium distribution of the conformational set provides a very good explanation of the experimental IR spectrum measured at T = 548 K.
The conformations of capped proline are searched by the method incorporating the four-atom coplanar rule of the pyrrolidine ring. In addition to the five conformations known previously, two new conformers of capped proline are found. The seven conformers fall into an energy range of 6 kcal mol−1. The capped proline conformations indicate that all P–Xnp peptide planes should adopt the trans form. The trans form restriction can be used to cut by half the cost of the conformational search of the proline-containing peptide, requiring only 48 combinations of bond rotations for a complete PES scan of a proline residue. The X–P peptide bond may adopt either the trans or the cis forms. The equilibrium compositions of the cis X–P peptide bonds for 98 K ⩽ T ⩽ 548 K have been presented. The theoretical composition of cis X–P peptide bonds at T0 is 5.5%, agreeing well with the experimental estimations of 3%–10%.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We are grateful for the financial support from the National Natural Science Foundation of China (grant nos. 12074362 and 11774324) and the computing time at the Supercomputing Center of the University of Science and Technology of China.