
Abstract
The effects of different substituents, located at the para position of the aromatic ring and at the β-carbon atom of styrenes, on difunctionalizations involving trifluoromethylation and oxime formation are investigated, showing that the difunctionalization reaction has a good adaptability to such reactants containing a range of substituents. This is important in the actual production process. It was found that proton transfer in the final tautomerism step involving transformation of a nitroso intermediate into an oxime is the rate-limiting step. The solvent effect did not influence the rate-limiting step significantly. Compared with direct proton transfer in a vacuum, the energy barrier of the final tautomerism step decreased from 57.80 kcal mol−1 in vacuum to 12.98 kcal mol−1 in water occurring via mediated proton transfer, which declines by 77.5%. When water participates in the rate-limiting steps in organic solvents, the energy barrier also decreases significantly, which indicates that a small amount of water in the organic solvent is conducive to the reaction.
Keywords

Highlights
The simultaneous trifluoromethylation and oximation of styrene was studied.
The solvent effect has little impact on the overall reaction.
The effect of substituents on rate-limiting steps is relatively small.
The energy barrier of the tautomerism decreased by H2O-mediated proton transfer.
Introduction
Fluorine is a substance with high electronegativity and activity. 1 Introducing fluorine into organic molecules can dramatically change their physical, chemical, and biological properties. 2 At present, there are mainly two types of methods for introducing a fluorine atom to an organic compound: direct C–F bond formation 3 and the fluorinated building block method. 4 The first method can be divided into three subtypes according to the electrical property of the reagent: electrophilic-fluorination, nucleophilic-fluorination, and radical-fluorination reactions. 5 Generally, most of these reactions are harsh and difficult to control. 6 The fluorinated building block route is an important method for synthesizing fluorinated organic compounds. It uses different fluorinated organic intermediates as fluorinated building blocks to synthesize fluorine-containing target molecules through appropriate reaction pathways.7,8 Compounds containing a trifluoromethyl group are often used as fluorinated building blocks in modern chemistry. 9 The introduction of a trifluoromethyl moiety into a substrate can significantly improve its biological activity.10–12 Some organic compounds containing trifluoromethyl groups play very important roles in pharmaceuticals, agrochemicals, and in organic materials. 13 Reactions involving the fluorinated building block method generally do not involve the breakdown and formation of a C–F bond, and benefit from mild reaction conditions, good selectivity, and high yields.14,15
The oxime structural motif is widespread in natural products16,17 and in many biologically active compounds.18,19 It is an important synthon for the syntheses of amines,20,21 amides, 22 nitriles,20,23 and heterocyclic compounds. 24 Although there are a variety of synthetic methods that have been developed for the preparation of oximes, they usually require the presence of a carbonyl or a nitro group in the substrate.25,26 Simultaneously introducing two functional groups to an aryl olefin can significantly simplify the reaction steps and shorten the preparation time, thus improving the atom economy and reducing waste. It has been reported that α-sulfonyl ketoximes can be prepared via the 1,2-difunctionalization of styrenes with sodium arylsulfinates and tert-butyl nitrite (TBN).27,28 Our group previously investigated the bifunctionalization reaction of aryl-substituted ethylenes with the Langlois reagent (CF3SO2Na) as trifluoromethylation reagent and TBN as the oxidant and oxime source, which introduced a trifluoromethyl and an oxime simultaneously at the double bond position. Based on nuclear magnetic resonance (NMR) and isotope-tracking experiments, the putative reaction mechanism shown in Scheme 1 was proposed. 29
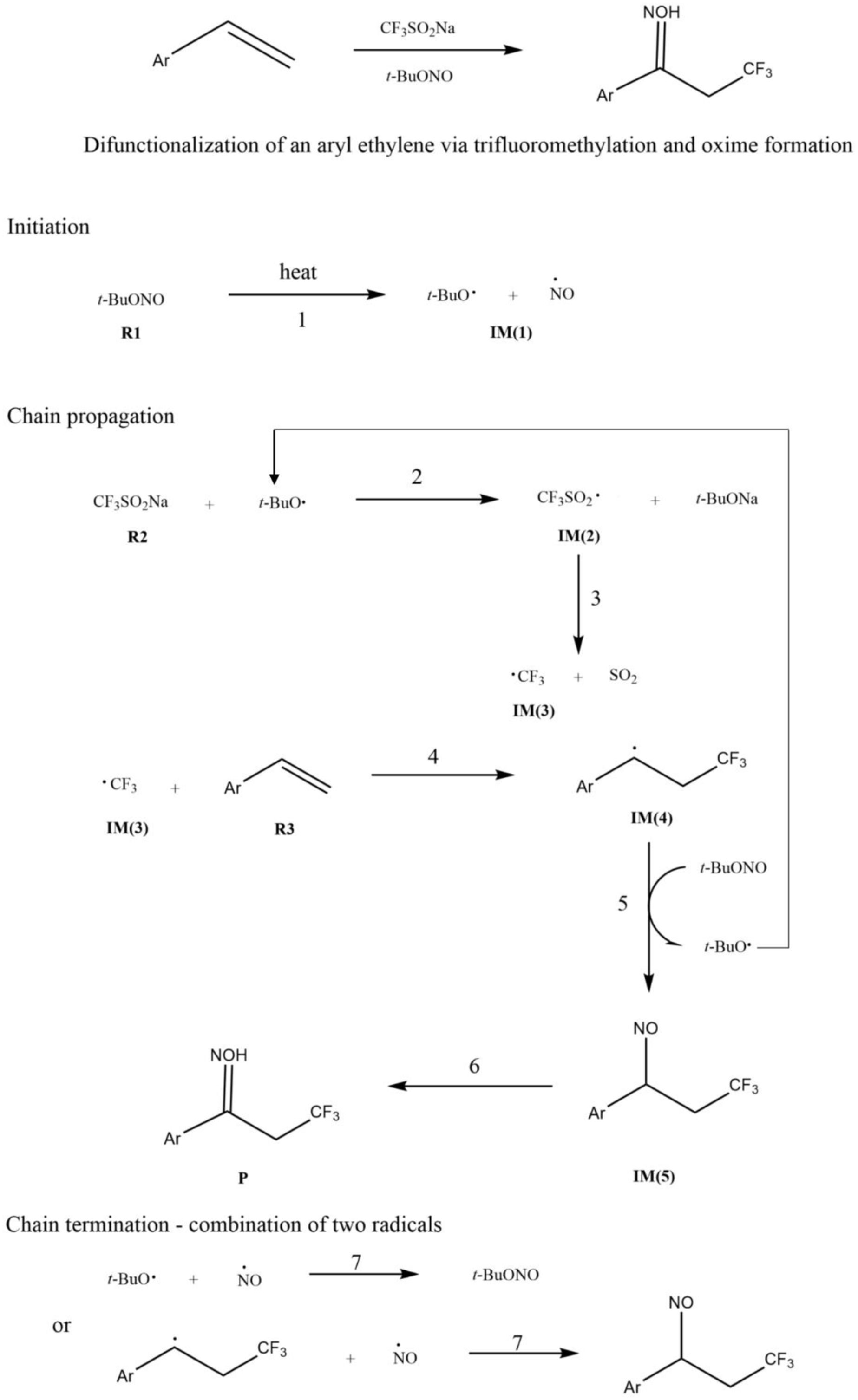
The reaction mechanism for the simultaneous trifluoromethylation and oximation of aryl ethylenes.
For novel chemical pathways, only when the reaction energy barrier, the reaction rate, the enthalpy change of each step, and the rate-limiting step of the whole reaction process are clearly known can new methods be readily applied to synthesis of other compounds. It is well known that substituents have a significant influence on reaction sites and reactivity.30–32 This is bad news for synthetic chemists in some circumstances. First, because of the influence of the existing substituents, it is difficult to determine where the subsequent substituents attack. Second, the rate at which a substituent attacks the desired reaction, site changes significantly as the surrounding substituents change. Shen et al. 33 investigated the activities of the electrophilic sites on the benzene ring of 4-substituted anilines and their acyl compounds through quantum chemical computations and chemical synthesis. Shiraz et al. 34 used NICS (negative nucleus-independent chemical shift) calculations and energy barriers (ΔE) to predict the most reactive site in azulene. It is common in chemistry to study the effects of substituents on reaction sites and reactivities by quantum chemistry computation. Solvent effects also have a great influence on chemical synthesis and the energy barrier and reaction rate constant of each step in a synthetic process. 35 In this study, quantum chemistry calculations were employed to examine the transition state, the energy change of each step of the reaction and the rate-limiting step. The impacts of substituents and solvent effects on the reactivity and reaction sites of aryl ethylenes during trifluoromethylation and oxime formation were also investigated, so as to predict the effects of different substituents and solvents on the reactivities of the radical reactions during simultaneous trifluoromethylation and oximation of aryl-substituted styrene derivatives, and to provide theoretical guidance for related reactions.
Results and discussion
The formation of trifluoromethyl radicals in vacuum
The proposed reaction process for the simultaneous trifluoromethylation and oximation of aryl-substituted ethylenes is given in Scheme 1. The whole reaction proceeds via a free-radical chain process. The difunctionalization reaction can be divided into three processes involving seven steps: (1) The first process consists of three steps: 1, 2, and 3. TBN forms a tert-butoxy radical and a nitric oxide radical on heating. Radical transfer from the tert-butoxy radical to the Langlois reagent (CF3SO2Na) forms a trifluoromethylsulfinic radical, which is followed by homolysis to give the trifluoromethyl radical. (2) The second process also includes three steps: 4, 5, and 6. The aryl-substituted ethylene reacts with the trifluoromethyl radical and TBN to give the α-trifluoromethylethylketoxime. (3) The third process consists of only step 7, and involves termination of the reaction via radical combinations; this step is not discussed later. The formation of trifluoromethyl radicals includes three steps and the optimized structure parameters of the reactants (R), transition states (TS) and intermediates (IM) in vacuum and in the dimethyl sulfoxide (DMSO), ethanol, and water are shown in Figures 1 and 2. The potential energy surface for the formation of trifluoromethyl radicals in vacuum is given in Figure 3.

Some optimized structural parameters of the reactant (

The optimized initial and transition state structures (

The potential energy surface of the difunctionalization reaction of an aryl-substituted ethylene in vacuum.
The bond length of N(6) to O(5) is 1.398 Å and the C(1)–O(5) distance is 1.481 Å in TBN (R1). The bond length of the nitrogen–oxygen double bond is 1.188 Å. The angle for O(7)–N(6)–O(5) is 110.909°. As the reaction proceeds, the N(6)–O(5) bond length increases leading to a transition state (
It can be seen from Table 1 that t-butyl nitrite has a transition state to dissociation and did not follow a Morse curve–type dissociation, which may be due to the reduction of energy caused by the conformational adjustment of the product of step 1. The energy barrier of the homolysis of TBN was 47.42 kcal mol−1, which was obtained from the relative energy difference between the transition state
The energy barriers (∆E), reaction enthalpy changes (

: in vacuum; D: DMSO (dimethyl sulfoxide); E: ethanol; W: water; –: not obtained.
The process of the trifluoromethyl radical attacking styrene to produce an α-trifluoromethylethyl ketoxime in vacuum.

The potential energy surface scanning of the distance between O(8) of the tert-butyl radical and Na(9) of sodium trifluoromethyl sulfonate in step 2 in vacuum. (a) Energy change in the potential energy surface scanning. (b) Molecular structure of a frame during scanning: DO(8)–Na(9): the distance between O(8) of the tert-butyl radical and Na(9) of sodium trifluoromethyl sulfonate (oxygen: red; sulfur: yellow; carbon: gray; fluorine: cyan; sodium: purple; hydrogen: white).
In this work, styrene was used as an initial representative of an aryl-substituted ethylene. It is synthetic resin monomer and an important industrial raw material for preparing ion exchange resins and synthetic rubber in industry. In addition, styrene can be used in pharmaceuticals, dyes, pesticides, and mineral-processing industries. 36 The trifluoromethyl group is often used as a bioisostere in place of chlorine or methyl for the preparation of various derivatives. It can be used to regulate the configuration and electronic properties of lead compounds or to protect active methyl groups from metabolic oxidation. 37 Trifluoromethyl radicals are commonly used to install trifluoromethyl functional groups. In this research, the addition of a trifluoromethyl group to the C=C double bond of styrene and subsequent reactions were explored.
The process of trifluoromethyl radical attack on styrene (Scheme 1) mainly has three steps: steps 4, 5, and 6. Step 4 involves attack of the trifluoromethyl radical (
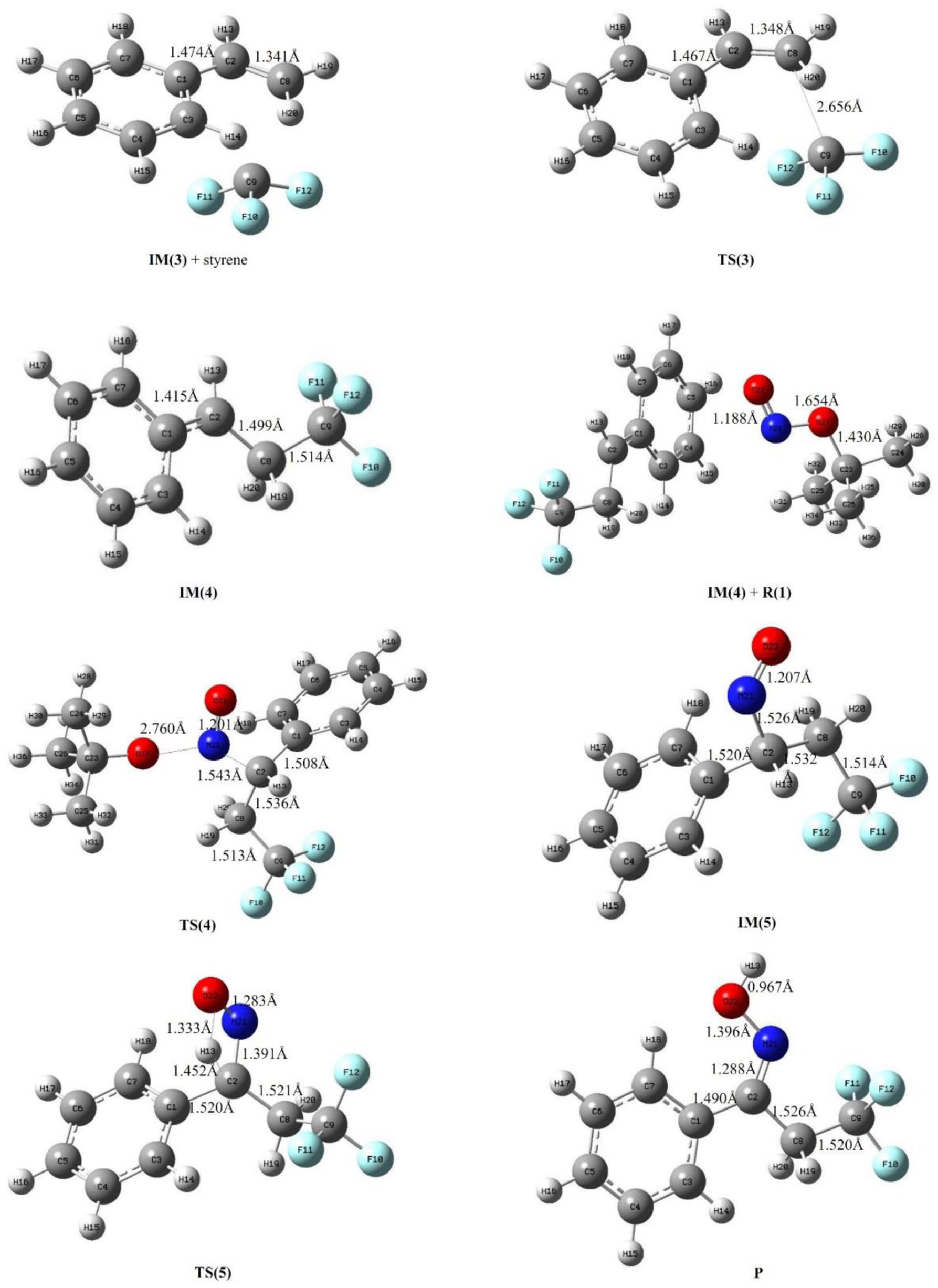
The optimized structures and some of the main parameters of the intermediates (
Effect of the para substituents of the aryl ring and the β-C substituents of styrene on the difunctionalization reaction in vacuum
The substituents on the aromatic ring of styrene can affect the electron density distribution on the benzene ring 38 and the conjugated vinyl bond, and further may affect the difunctionalization reaction. Grainger et al. 39 reported that ortho substituents close to the reaction sites may have some influence on the formation of transition states. Electron-donating and the electron-withdrawing substituents have different effects on the electron distribution of the reactants and the transition states, and further influence the energy barrier of the reaction. 40 To examine the effect of para substituents on the aryl ring on the difunctionalization reaction, the difunctionalizations of the para-substituted styrene with electron-donating methoxy and methyl groups and an electron-withdrawing of fluorine group were examined in this study. To further explore the effects of substituents on the difunctionalization reaction, the native of the β-carbon substituents were also investigated. We chose methoxy and fluorine as representatives of electron-donating and electron-withdrawing groups, respectively, on the β-C. Since the first process only produces a trifluoromethyl radical and does not involve the subsequent reaction of styrene, the substituents only affect the second process. Hence, steps 4, 5, and 6 of the difunctionalization reaction with the substituted styrene were examined. Tables 2 and 3 list the energy barriers that needed to be overcome and the reaction rate constants for steps of 4, 5, and 6. Figure 6 shows the optimized structures and some main parameters of the intermediates, transition states and products of steps 4, 5, and 6 of the difunctionalization reaction of para-substituted styrene at the B3LYP/6-31+G(d,p) level in vacuum. The corresponding structural parameters of the intermediates, transition states and products in steps 4, 5, and 6 are listed in Figure 7.
The energy barriers (∆E4, ∆E5, and ∆E6) and reaction rate constants (k4, k5, and k6) of steps of 4, 5, and 6 for the difunctionalization of para-substituted styrenes in vacuum (V) and in different solvents: dimethyl sulfoxide (DMSO (D), ethanol (E), and water (W).

The energy barriers (∆E4, ∆E5, and ∆E6) and reaction rate constants (k4, k5, and k6) of steps of 4, 5, and 6 for the β-C substituents on styrenes in vacuum (V) and in different solvents: dimethyl sulfoxide (DMSO (D), ethanol (E), and water (W).

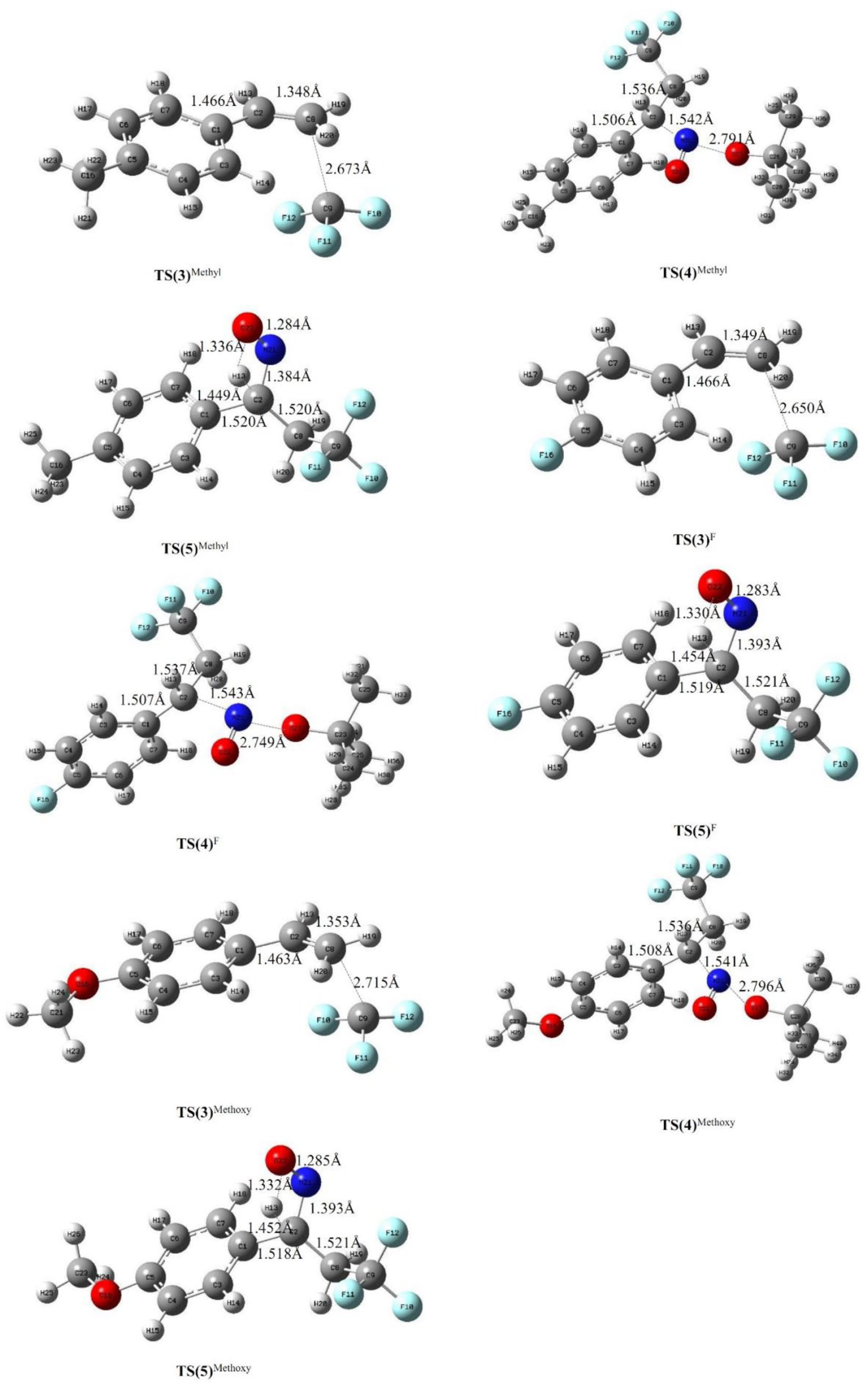
The optimized structures and some of the main parameters of the intermediates (
The optimized structures and some of the main parameters of the intermediates (
Table 2 shows the energy barrier change of the second process with the different para substituents on the aryl ring of the styrene. When the substituent was an electron-donating CH3O group at the para-position, the reaction energy barrier of step 4 directly decreased from 24.84 kcal mol−1 (H) to 0.03 kcal mol−1 (–CH3O), a decrease of 24.81 kcal mol−1, and the reaction rate constant went from 1.952 × 10−12 s−1 M−1 to 3.242 × 106 s−1 M−1. However, an electron-withdrawing F substituent increased the energy barrier of step 4 by 5.50 kcal mol−1 in vacuum. Among all the substituents, a methoxy group had the greatest effect on step 5, increasing the reaction energy barrier by 0.51 kcal mol−1 and the reaction rate constant reduced from 8.073 × 103 s−1 M−1 to 7.950 × 103 s−1 M−1. It can be seen that the substituents only have a small effect on step 5, though both electron-donating and electron-withdrawing groups decreased the reaction energy barriers. For step 6, both electron-donating and electron-withdrawing substituents marginally increased the energy barrier in vacuum, and the maximum increase was only 0.30 kcal mol−1 in the presence of a methoxy group. This is consistent with the reported experimental results. 29 The substituents have little influence on the yields of the final products (α-trifluoromethylethyl ketoxime). Hafner and Bräse 41 demonstrated that triazenes para electron-withdrawing substituents had little effect on the yield of trifluoromethyl products, which is also consistent with the effect of para substituents on the trifluoromethylation of aryl compounds found in this paper.
Comparing the data in Table 3 with that in Table 1, when a methoxy group is located at the β-carbon, the reaction energy barrier of steps 4, 5, and 6 is increased by 3.11 kcal mol−1, 0.03 kcal mol−1, and 0.45 kcal mol−1, respectively, in vacuum. Fluorine, as the β-C substituent, has a greater effect on the energy barrier, increasing the energy barriers of step 4 by 6.43 kcal mol−1 and reducing the barrier of step 5 by 2.39 kcal mol−1. For the rate-limiting step, an F substituent located at the β-carbon reduces the reaction barrier by only 0.80 kcal mol−1. We know that the rate-limiting step of the reaction plays a decisive role in the reaction. As a whole, for para and β-carbon substituents, neither the electron-withdrawing nor the electron-donating substituent has much effect on step 6, indicating that the difunctionalization reaction has good applicability for substituted styrene derivatives with different substituents at the para and β-carbon positions, and thus can be applied to the synthesis of many differently substituted styrene derivatives.
The effect of different solvents on the difunctionalization reaction
Methods for controlling chemical reactions have always been a goal in chemistry. The solvent effect is a very important aspect in chemical reactions and is usually used to control the speed and selectivity of a reaction.42,43 Therefore, it is necessary to obtain the theoretical data of a reaction in different solvents so as to guide experiments. To examine the solvent effect on the difunctionalization of substituted styrenes, the reaction processes in DMSO, ethanol, and water as the solvents were examined at the B3LYP/6-31+G(d,p) level with the CPCM (Conductor Polarized Continuum Model) solvent model. For unsubstituted styrene, the energy barriers for the homolysis of TBN in DMSO, ethanol, and water were 47.88 kcal mol−1, 47.86 kcal mol−1, and 47.88 kcal mol−1, respectively (given in Table 1), which were 0.46 kcal mol−1, 0.44 kcal mol−1, and 0.46 kcal mol−1 higher than that in vacuum. The reaction rate constants for the homolysis of TBN are 3.558 × 10−24 s−1, 3.680 × 10−24 s−1, and 3.558 × 10−24 s−1, respectively. In this section, the subsequent value orders given correspond one-to-one to the solvent order with the former in DMSO, the middle in ethanol, and the latter in water. The optimized structures of the reactants, transition states, and intermediates of the first process are similar to the data presented in vacuum. It was found that the presence of a solvent was not conducive to the homolysis of TBN because the energy barriers only changed marginally. Step 2 is still a barrierless process in the solvent (potential energy surface scans are not provided), and their reaction enthalpy changes are 27.35 kcal mol−1, 27.27 kcal mol−1, and 27.36 kcal mol−1, respectively. Subsequently, the generated CF3SO2· radical was homolyzed to give ·CF3 and SO2. The energy barriers for the breakage of the C–S bond are 4.97 kcal mol−1, 4.99 kcal mol−1, and 4.96 kcal mol−1, respectively. The energy barriers in the third step are 0.71 kcal mol−1, 0.69 kcal mol−1, and 0.72 kcal mol−1 lower than that in vacuum. In the solvents, the reaction rate constants are 9.859 × 107 s−1, 9.531 × 107 s−1, and 1.002 × 108 s−1, respectively. The optimized structures of the intermediates, transition states, and products in the second reaction process of the difunctionalization of styrene are very similar, so only the structure diagrams in DMSO are given in Figure 8. By comparing the energy barrier of step 4 in vacuum with those in solvents, we found that the barriers in DMSO and ethanol are 0.75 kcal mol−1 and 0.74 kcal mol−1 lower than that in vacuum. However, in water, the energy barrier increases by 8.80 kcal mol−1. In the presence of solvents, the reaction energy barriers in step 5 are increased by 1.31 kcal mol−1, 1.27 kcal mol−1, and 1.33 kcal mol−1, respectively. The energy barriers of the rate-limiting step (step 6) in the solvents are all 58.36 kcal mol−1 and the rate constants are all 3.771 × 10−31 s−1. From the Gibbs free energy change (

The optimized structures and of the some main parameters of the intermediates (
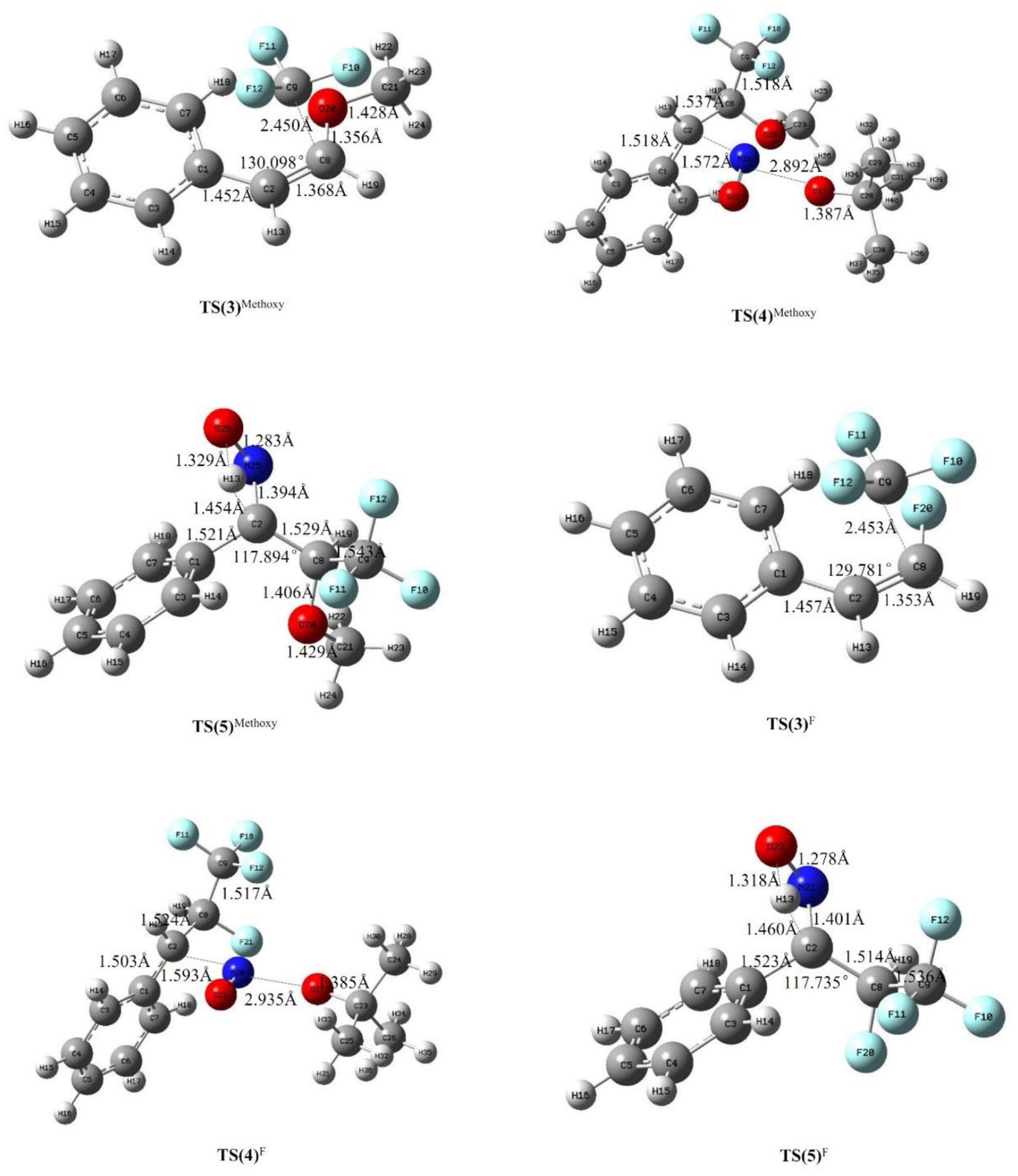
The solvent effect on the reactions with substituted styrenes as reactants was also examined. Figures 9 and 10 show the optimized structures of the transition states of the substituted styrenes in the second reaction process of the difunctionalization in DMSO as the solvent. In other solvents, the optimized structures of the transition states are very similar to that in DMSO and are not listed here. The energy barriers and reaction rate constants of all the steps in the solvents are listed in Tables 2 and 3. It can be seen that the solvents slightly increase the energy barriers of steps 4 and 6 and decrease that of step 5. This trend did not change with the position and type of substituents on the styrene.

The optimized structures and some of the main parameters of the transition states (

The optimized structures and some of the main parameters of the transition states (
In general, the solvent has little effect on the overall reaction process. Zhao et al. 44 found that the solvent also had little effect on the yields of the trifluoromethylselenolation of indole and the trifluoromethylthiolation of 4-aminobenzoate. 45 However, in solution, the molecules of the reactants can be dissolved and are in sufficient contact to allow the reactions to proceed smoothly. DMSO has good solubility for most of the organic reactants and it is helpful that the reactants are sufficiently mixed for the reaction to occur. On the contrary, the energy barrier of the final step of the reaction in this study is as high as 57.80 kcal mol−1 and the energy barrier of step 1 is 47.42 kcal mol−1 in vacuum, indicating that this reaction should be carried out at a higher temperature, and the high boiling point of DMSO can provide a higher temperature environment to facilitate this reaction. Therefore, the actual reaction is chosen to be carried out in DMSO as the solvent. 29
Water-mediated proton transfer in the oximation reaction
In the final step (step 6), the oxime product was formed via tautomerism of the nitro compound (such as

The structural parameters and energy barriers of the transition states (
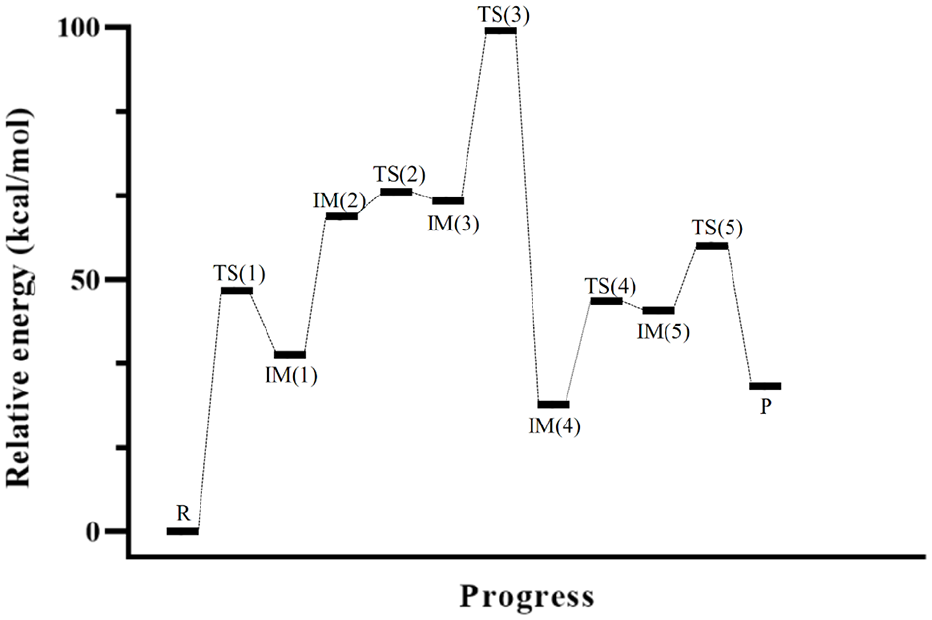
The potential energy surface for the proton transfer mediated by two water molecules in the solvent (water).
Conclusion
In this paper, the free-radical reaction mechanism of the difunctionalization of aryl-substituted ethylenes has been investigated by using the Gaussian09 package at the B3LYP/6-31+(d,p) level in vacuum and the solvent effects of DMSO, ethanol, and water were examined with the CPCM solvent model. It was found that step 2 involving formation of CF3SO2·(
Computational methods
All of the quantum chemistry computations and structure optimizations of the reactants, products, and the transition states are based on the density functional theory (DFT).
53
All the computations were performed with the Gaussian09 package.
54
All molecular geometries including those of the reactants, intermediates, and products in the reactions in vacuum and in solvents (DMSO, ethanol, and water) were optimized at the B3LYP/6-31+G(d,p) level. The solvent effects were simulated using the CPCM model.
55
Unrestricted spin calculations were used for all the open-shell systems. The TS and QST2 methods were used to find the transition states when the structures of the reactant and the product in the reaction were fully optimized. Frequency analyses were conducted to determine whether a given structure is a minimum or a transition state. Calculations of intrinsic reaction coordinates (IRC) were employed to confirm the located transition states, which connect with the expected minima. All initial states, stable intermediates, and final products were confirmed to have only real harmonic frequencies and each transition state was confirmed to have one single imaginary vibrational frequency.
48
The energy barrier of the reaction was obtained from the energy difference between the transition states and the corresponding reactants with the zero-point correction. The reaction enthalpy change (

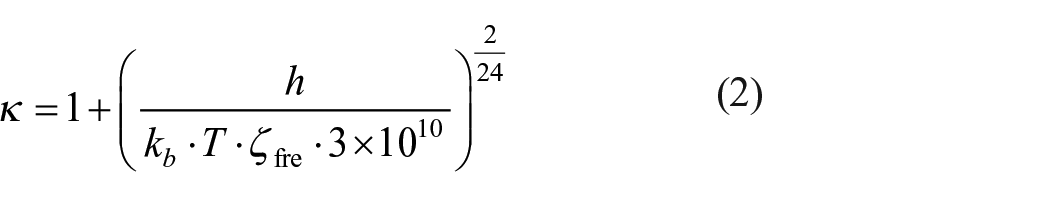

In the above equation,
Supplemental Material
sj-docx-1-chl-10.1177_17475198221104006 – Supplemental material for A density functional theory study on the mechanism of simultaneous trifluoromethylation and oximation of aryl-substituted ethylenes
Supplemental material, sj-docx-1-chl-10.1177_17475198221104006 for A density functional theory study on the mechanism of simultaneous trifluoromethylation and oximation of aryl-substituted ethylenes by Sen Wang, Wenlu Song, Xiaowei Lan, Xuan Meng, Nan Li, Xianfu Wei, Wenjie Jing, Kui Lu and Yujie Dai in Journal of Chemical Research
Footnotes
Acknowledgements
We thank Professor Yujie Dai for his language help, writing assistance, and proofreading this paper.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of Tianjin (grant number 19JCZDJC34800) and the COVID-19 Prevention and Control Project of Tianjin University of Science and Technology (grant number 2020STCV0016).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.