
Abstract
The functionalisation of two-dimensional materials is key to modify their properties and facilitate assembly into functional devices. Here, new reactions have been proposed to modify crystalline two-dimensional carbon nitrides of polytriazine imide structure. Both amine alkylation and aryl-nitrene-based reactions have been explored computationally and with exploratory synthetic trials. The approach illustrates that alkylation is unfavourable, particularly at basal-plane sites. In contrast, while initial trial reactions were inconclusive, the radical-addition of nitrenes is shown to be energetically favourable, with a preference for functionalising sheet edges to minimise steric effects.

Introduction
Two-dimensional (2D) nanomaterials that are already well known for their remarkable properties arising from quantum confinement effects, large surface areas and aspect ratios are now attracting significant interest due to their possibilities for surface functionalisation and reactivity. 1 One class of materials being developed for such applications includes layered polymeric or ‘graphitic’ carbon nitrides that are being studied extensively for redox catalysis, with a bandgap around 2.7 eV enabling photocatalytic reactions including water splitting, and reactive sites associated with both Lewis and Brønsted acid–base functionalities.2–6
Crystalline carbon nitrides of discrete 2D sheets are known, most notably in the form of polytriazine imide (PTI). Bulk PTI samples are produced by addition–elimination reactions between organic precursors (e.g. melamine, cyanuric chloride) or by condensation of dicyandiamide (DCDA: C2N4H4) in molten alkali halide salt mixtures.7–10 The PTI crystalline structural motif consists of 1,3,5-triazine rings linked by imide (–NH) bridges with voids appearing in the layer plane. 11 In the solid state, the 2D sheets, whose unit cell has composition of C6N9H3, stack to form slightly buckled layers in either an AA′ or AB fashion. In AA′ stacking, the triazine rings sit above and below one another and in AB stacking the structural voids sit above and below the triazine rings of adjacent layers. 12 As initially synthesised, these crystalline compounds typically contain intercalated ions (e.g. Li+, Cl−, Br−) derived from the precursors or synthesis medium, and these ions can be exchanged or completely removed without loss of crystallinity or disruption of the layered structure.13–15 It was recently shown that the PTI layers can be exfoliated and dissolved spontaneously to form true solutions or nanoparticle dispersions by simply placing them in contact with aprotic solvents (dimethyl sulfoxide (DMSO), dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP)) without agitation.12–14,16 Combining high-resolution transmission electron microscopy (TEM) and rapid scanning atomic force microscopy measurements on nanomaterials re-deposited on surfaces, along with photoluminescence (PL) studies comparing nanoparticle solutions with re-deposited solids, showed that the PTI-structured carbon nitrides retained the ordered arrangement of C and N atoms present in the original crystalline phase and that they formed few-layered luminescent species in the solution phase.9,13 Further experiments have demonstrated that at low concentration only very few-layered species are present in solution, while also multi-layered units are present as the concentration is increased. 12
The covalent attachment of functional groups to graphene and related nanostructures is used to tune their properties including (opto)electronics, dispersibility and reactivity for applications in fields ranging from catalysis to biomedicine.5,17–19 One report of covalent functionalisation of PTI used reductive charging followed by alkyl halide addition, based on previous work for single-walled carbon nanotubes. 20 While successful, the process relies on difficult to handle air-/water-sensitive reagents and is incompatible with functional groups sensitive to strong reducing agents, leading to a need to develop additional PTI functionalisation reactions. In the present work, we investigated (a) the methylation of and (b) organo-radical addition to the amine groups (–NH bridges and –NH2 edge sites) of PTI nanolayers as potential routes for surface/edge functionalisation in solution. We combined density functional theory (DFT) calculations with experimental trials to examine the thermodynamics of the proposed reactions. Our first investigations focused on methyl iodide (CH3I) that is a common methylating reagent for amine groups, leading to eventual formation of quaternary ammonium salts.21,22 Later experiments and calculations studied azidobenzene that degrades to form nitrene radicals via thermolytic or photolytic initiation and reacts strongly with amines to form azepines at reasonable yields and high selectivity.23,24 Derivatives of azidobenzene have previously successfully modified the basal plane of graphene with polyethylene glycol-substituted functionalities rendering the functionalised nanomaterial water soluble, enhancing its processibility.25,26
Results and discussion
Methylation reaction: DFT results
We investigated the methylation reaction using low dimensional models of the PTI structure to represent the exposed surface sites accessible to the CH3I reactant (Figure 1). These include a 2D monolayer describing the predominant (001) surface, that is, the natural layering orientation of the PTI structure, and a 1D polymer (or ribbon) section of the (001) plane extending along the (100) direction, used to represent the edges of the platelets. The C–N bonds cleaved upon formation of the 1D ribbon have been saturated and only expose –NH2 terminal groups. The 1D model employed had a thickness along the non-periodic direction corresponding to five undisrupted PTI voids, or 45 Å between edge –NH2 terminal groups on either side. These models contain two types of N-H bonds that can be functionalised: the –NH imide bridges oriented towards the centre of the C12N12H3 triangular voids that make up the (001) plane, and the −NH2 edges of the 1D ribbon. A 2 × 2 supercell expansion of composition C24N36H12 was used in the 2D model to create structures with different degrees of methylation at the –NH bridge sites. In the 1D ribbon, structures with alternating edge functionalisation were simulated using a 2× supercell expansion along the periodic a direction, with overall composition C36N55H21.

Low-dimensional models of the PTI structure: (a) 2D monolayer representing the (001) surface of bulk PTI and (b) 1D polymer (or ribbon) extending along the (100) direction, obtained from the 2D (001) plane by cleaving along the dashed red lines. The C–N bonds cleaved are saturated, so that they expose NH2 groups at the edge. The models contain two types of accessible –NH bonds: the –NH imide bridges in the C12N12H3 triangular voids that make up the basal plane, indicated by the blue circle, and the –NH2 sites that define the edges of the 1D model, indicated by a red circle. The unit cells are defined by parallelogram ab in the 2D model and line a in the 1D model.
We have considered the reactive attachment of CH3I to the N atoms in the –NH bridges and at –NH2 edges (Figure 2). Each PTI void has three –NH imide bridges available for methylation. Our calculations started by assuming a single functionalised site in one-fourth of the voids, then increasing the concentration to two sites in one-fourth of the voids, a single site in half of the voids, a single site in three-fourths of the voids, two sites in each void and finally to full methylation. This series allowed us to investigate structural and energetic variations associated with multiple methylation steps, either in the same or adjacent C12N12H3 voids of the PTI structure. In the 1D ribbon model, in which the 2× cell employed contains four –NH2 edge sites, structures with one to four methyl substituents were investigated, considering functionalisation on one only or both sides of the ribbon.

Scheme for the methylation of the –NH bridges in PTI with CH3I.
Subsequent methylation steps, on increasing concentration of the functionalised bridges, are represented by the following reaction
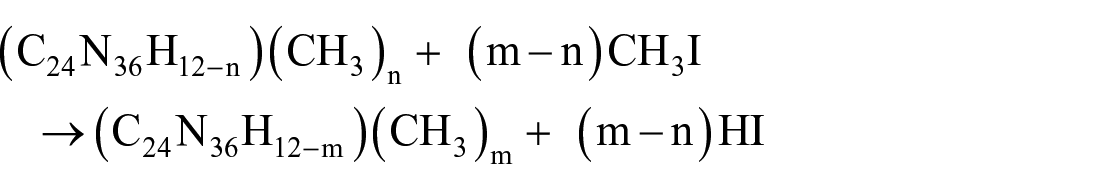
using the composition of the 2 × 2 two dimensional cell, where n and m represent the initial and final number of CH3 groups. The equivalent reaction for the 1D ribbon using 2× cell expansion is given in the following

The associated energy change for subsequent methylation steps at the bridge sites was calculated using the equation

whereas the energy change required for stepwise methylation of edges was estimated using the following equation
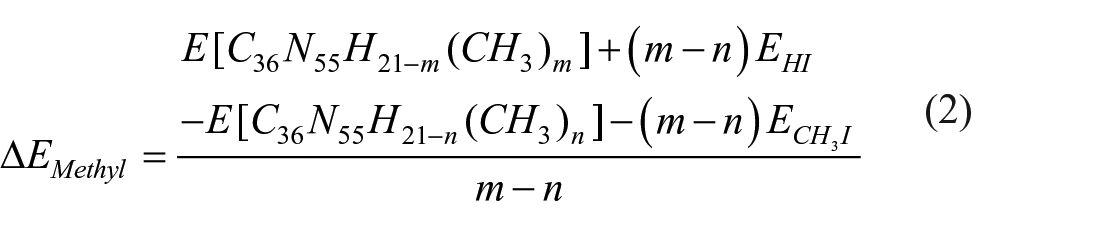
where energies of the hydrogen and CH3I species implied in the reaction were calculated for individual molecules in vacuum.
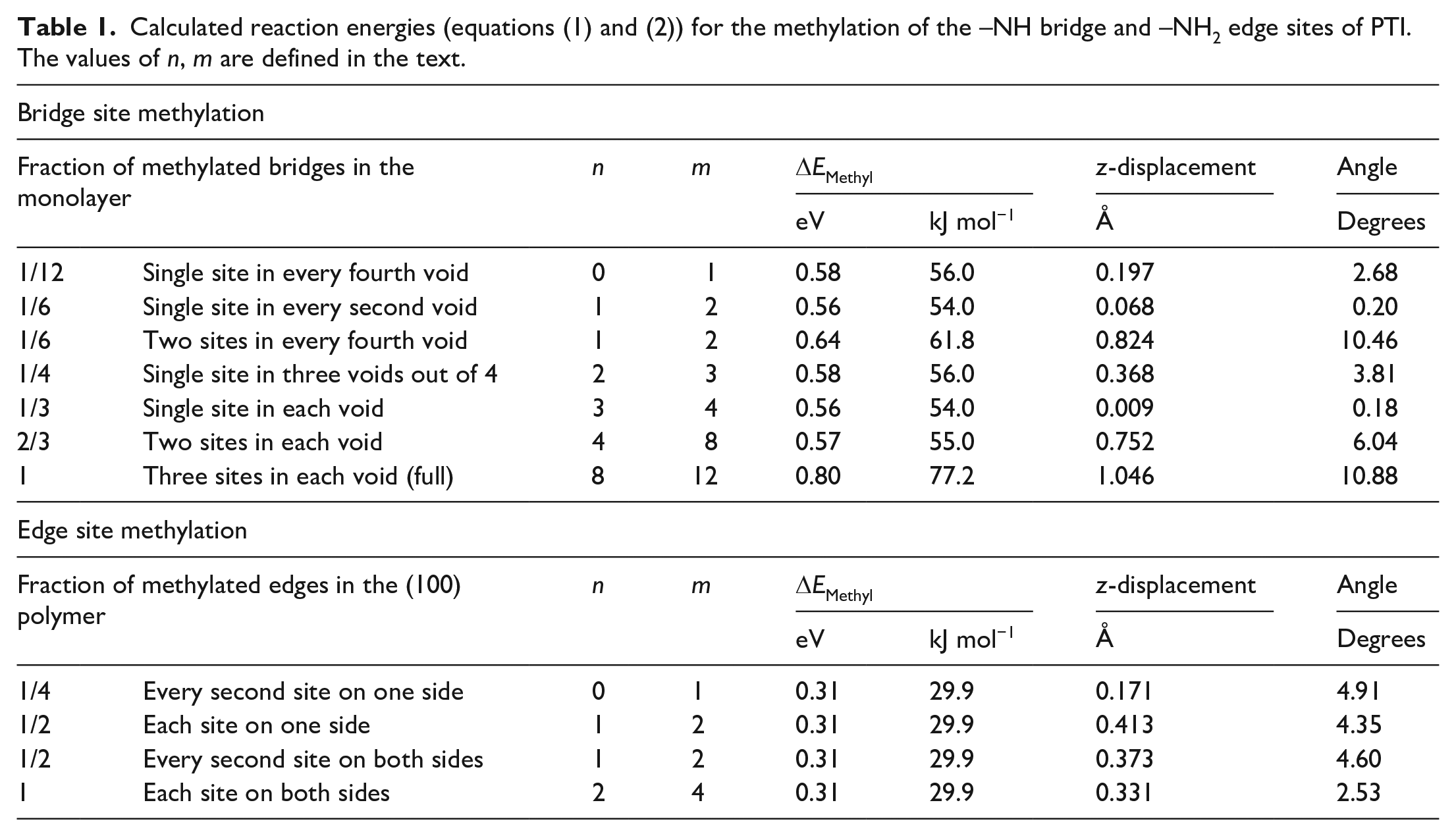
Calculated reaction energies, methyl out-of-plane displacement, and triazine-N-Me torsional angles associated with different degrees and modes of methylation of the bridge and edge sites are given in Table 1. The positive energy values seen for all modelled systems indicate that the methylation reactions were always endothermic and are not expected to proceed at any temperature; since the reaction has the same number of reactant and product molecules, the entropic contribution is not expected to offset the enthalpy change. Results clearly indicated that the edge sites are more reactive than the imide bridges, an observation that we associate with the more open structure around the edge –NH2 groups. This is further supported by the fact that the degree of functionalisation of the edge sites has no effect on energy attributed to the lack of additional disruption to the PTI structure after the first methylation, as seen in the similar out-of-plane displacements and triazine-methyl angles (Table 1). In contrast, methylation of the intralayer imide bridges not only is more endothermic but also increases with the number of methyl groups within a single void (Figure 3(a)). Methylation of two adjacent voids, instead, has no bearing on the calculated energies indicating that each void of the (001) PTI surface is in practice independent. When three-quarters of the voids are mono-methylated, the energy is comparable to one-quarter of the voids being mono-methylated, attributed to the similar levels of disruption to the PTI framework, reflected in the similar out-of-plane displacement of the methyls (Figure 3(b) and (c)). Top and side views of the methylated (001) surface are given in Figure 4. Throughout these methylation reactions, the PTI monolayer remains planar; however, when two (Figure 4(c), (g), (j) and (m)) or three (Figure 4(k) and (n)) methyl groups occupy the same void the distortion of the layer is evident, indicating the occurrence of significant steric effects; this is consistent with the increased reaction energy reported in Table 1, from 0.58 to 0.64 to 0.80 eV for 1, 2 and 3 groups in the same void, respectively. Upon the first functionalisation in each void, the C of the methyl group is in the plane of the PTI layer (Figure 4(l)); when the void contains two methyl rings, they are displaced 0.752 Å out of the PTI plane (Figure 3(b)), and orient on opposite sides of the layer to minimise steric repulsion (Figure 4(m), minimum H-H distance 2.713 Å). The distortion occurs to a much larger extent when all three imide bridges in the same void are methylated (Figure 4(n), minimum H-H distance 2.197 Å), with a larger out-of-plane displacement (1.046 Å). Similarly, at the same total one-sixth degree of methylation, that is, 2 of the 12 supercell bridges methylated, when both methylations occur in the same void versus in separate voids the reaction energy (Figure 3(a), 62/54 kJ mol−1, respectively) and out-of-plane methyl displacement (Figure 3(b), 0.068/0.824 Å, respectively) are significantly higher. No significant distortion has instead been observed for the edge sites of the 1D ribbon, at all methyl concentrations examined (Figure 4(o)–(x)).
Calculated reaction energies (equations (1) and (2)) for the methylation of the –NH bridge and –NH2 edge sites of PTI. The values of n, m are defined in the text.
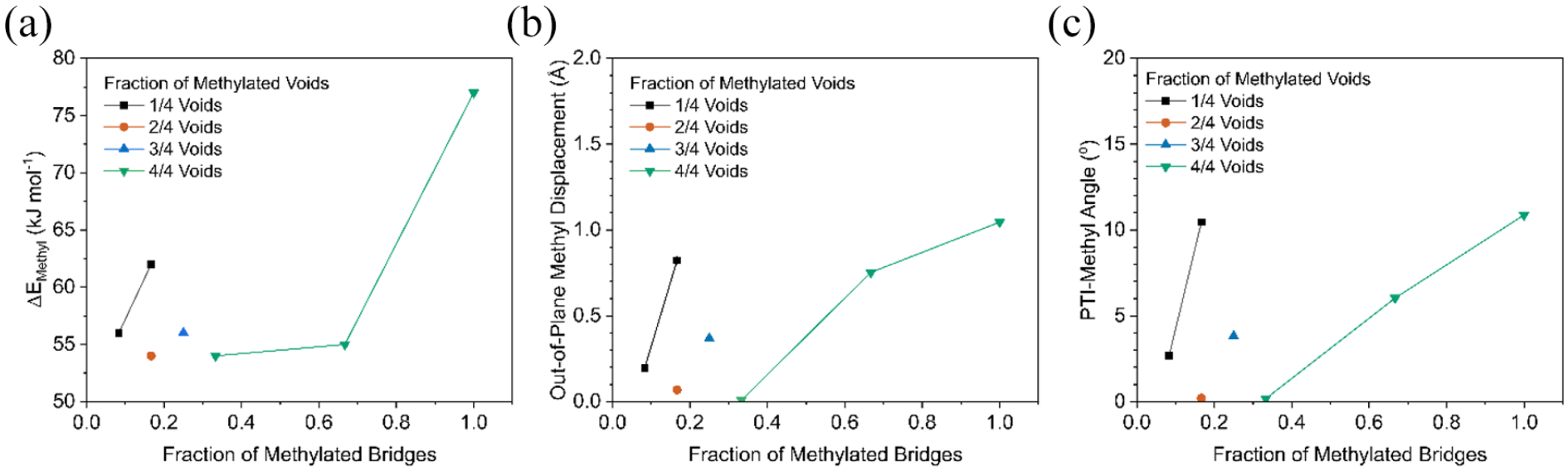
Formation energy and methyl structural properties of bridge-site methylated PTI versus fraction of methylated bridges, plotted separately by the fraction of voids which contain methylated bridges: (a) reaction energies, (b) z-axis displacement of methyl carbon versus average PTI-layer atom and (c) average torsional angle between methyl carbon and adjacent triazine units.
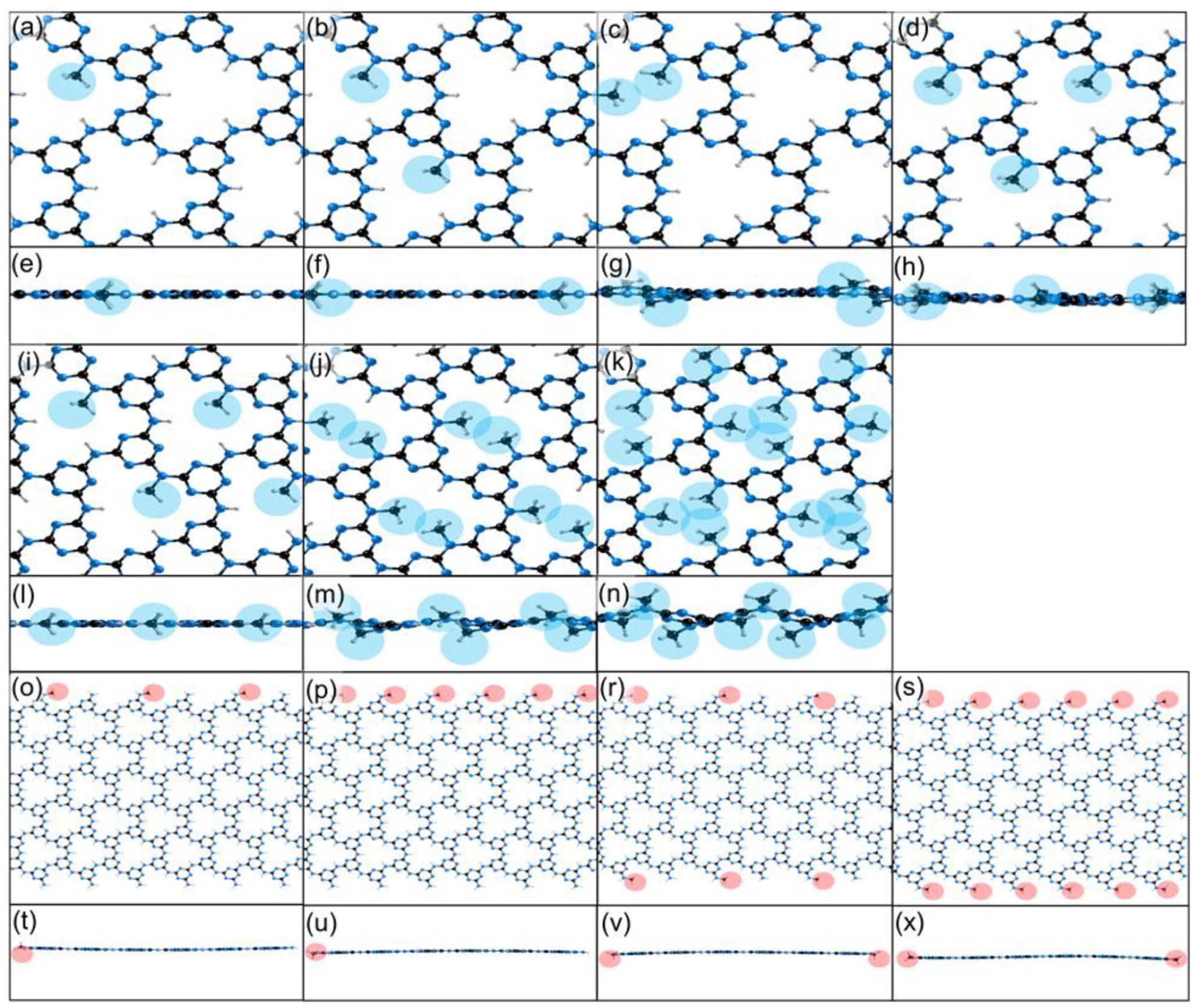
Optimised structures resulting from methylation of the –NH bridges (highlighted in blue) and –NH2 edges (highlighted in red) in the PTI monolayer, viewed along the (001) and (110) planes. The functionalised sites in each picture are represented by red or blue circles, with the same notation as in Figure 1. Bridge functionalisations. (a, e) 1/12 occupancy, single site in every fourth void; (b, f) one-sixth occupancy, single site in every second void; (c, g) one-sixth occupancy, two sites in every fourth void; (d, h) one-fourth occupancy, single site in three voids out of four; (i, l) one-third occupancy, single site in each void; (j, m) two-third occupancy, two sites in each void; (k, n) full occupancy, three sites in each void. Edge functionalisations (o, t) one-fourth occupancy, every second site on one side; (p, u) half occupancy, each site on one side; (r, v) half occupancy, every second site on both sides; (s, x) full occupancy, each site on both sides.
Methylation reaction: experimental results
The experimental trials supported the DFT results, in that the attempted methylation of PTI with CH3I was unsuccessful. No new peaks appear between wavenumbers 2800–3000 cm−1 in the FTIR spectrum of the product and the X-ray diffraction (XRD) pattern indicated that the PTI network remained unchanged (Figure 5). The XRD pattern for the PTI starting material differs from published literature on intercalant free PTI and instead closely matches intercalated PTI, suggesting that not all the potassium salt was removed by Soxhlet extraction. Small additional sharp peaks in the X-ray pattern correspond to K2CO3 remaining from the methylation reaction. 12

(a) FTIR spectra and (b) XRD patterns for CH3I-treated PTI in comparison to untreated PTI. The assigned Miller indices are in brackets and the red asterisks indicate impurity reflections from residual K2CO3 reactant.
Organo-radical addition: DFT results
The organo-radical addition of azidobenzene and 1-azido-4-fluorobenzene to PTI was investigated as a promising functionalisation alternative to methylation. Two possible mechanisms were proposed for the reaction (Figure 6). The thermal degradation of azidobenzene (X = H) to nitrene is known to undergo ring expansion, forming ketenimine, which would react with the –NH bridges and –NH2 edge sites to decorate the surface of PTI with azepine functionalities. 27 Halogenation of azidobenzene has been shown to raise the energy barrier for ring expansion and increase the lifetime of the halogenated nitrene radical. 28 Fluorination of the aryl ring in 1-azido-4-fluorobenzene (X = F) was thus hypothesised to suppress the ring expansion mechanism, resulting in the thermal degradation to 4-fluoronitrene and its direct reaction with the –NH bridges and –NH2 edge sites of PTI.

Scheme of organo-radical addition to PTI with azidobenzene (X = H) and 1-azido-4-fluorobenzene (X = F).
We used DFT calculations to study the thermal degradation of nitrene from azidobenzene and 1-azido-4-fluorobenzene via the reaction
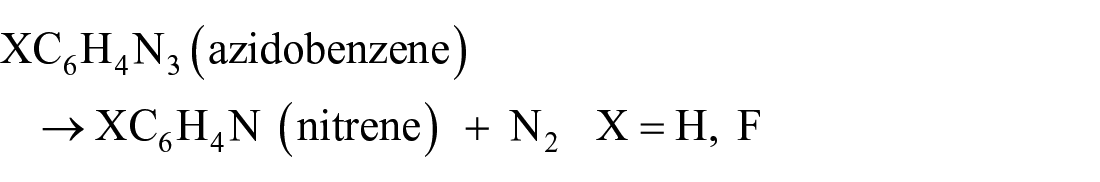
and calculated the associated energy change as
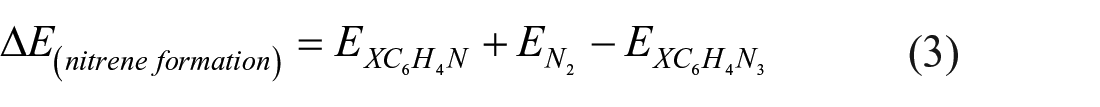
The calculated value of
Calculated reaction energies for the formation of nitrene and its ring expansion to ketenimine.

The energy difference between ketenimine and nitrene radicals (equation (4)) was used to represent the relative stability of the two decomposition products

Results reported in Table 2 showed that the ring expansion is associated with an energy penalty of 0.62 eV, that is, ketenimine was less stable than the nitrene radical. Changing the substituent from hydrogen to fluorine has no effect on the energy associated with the ring expansion. The thermal decomposition of azidobenzene is well documented in the literature; the energy penalty of ~0.2 eV calculated here for formation of the nitrene diradical does not rule out the azidobenzene decomposition at high temperature, especially considering that the reaction proceeds with elimination of N2 hence is accompanied by an entropy gain. 27 The formation of ketenimine through ring expansion is instead endothermic and is not affected by the fluorine substituent.
As for the methylation reaction, also for the organo-radical addition of azidobenzene to PTI we have employed the 2D (001) slab and 1D (100) ribbon models to represent and discriminate the reactivity of imide bridges and terminal −NH2 groups. Both the nitrene and ketenimine forms of the decomposition product have been investigated. Since steric effects were evident for multiple functionalisation of the imide bridges within the same PTI void even with the small methyl group, only a single functionalised bridge per PTI void (i.e. one-third of all possible sites) has been investigated here in the case of the 2D (001) surface. A variable level of functionalisation has been considered for the 2× supercell of the 1D ribbon, using the same geometries employed in the methylation. In general terms, the reaction is indicated as

where PTI-Ha is the composition of the (001) monolayer or (100) ribbon prior to functionalisation, n the initial and m the final number of PTI sites functionalised, with a corresponding reaction energy indicated as
The calculated reaction energies for both 2D and 1D models, nitrene and ketenimine forms, and at all concentrations are summarised in Table 3. In contrast to the energies calculated for the methylation reaction, here they all have negative values indicating the viability of the organo-radical reaction as a functionalisation mechanism for PTI. However, this reaction must be preceded by the formation of unstable nitrene or ketenimine reactant species.
Calculated reaction energies for the organo-radical addition to the –NH bridge and –NH2 edge sites of PTI.

We note in Table 3 that the calculated reaction energy for the ketenimine form is much more exothermic than for the nitrene; this is a result of the more unstable nature of the ketenimine intermediate (0.62 eV, see Table 2) that makes it more reactive. Addition to the imide bridges of PTI (2D model of Table 3) stabilises the ketenimine product compared to the nitrene by 0.14 eV (reaction energy of −1.41 compared to −1.27 eV); hence, even after interaction with the PTI surfaces, the nitrene form remains the most stable product of the azidobenzene decomposition.
The stabilising effect of ketenimine compared to nitrene upon addition to the −NH bridge has a structural explanation. The optimised structures arising from the organo-radical addition to −NH bridges of the PTI monolayer are shown in Figure 7. Addition to PTI results in bent geometries which orient the nitrene and ketenimine units away from the basal plane of PTI, to avoid steric repulsion from the carbon nitride framework. The seven-membered ring of ketenimine is no longer aromatic; hence, the loss of planarity is achieved already in the molecular intermediate and is retained in the solid product (Figure 7(a) and (c)). However, the nitrene molecule retains the planar aromatic structure of the aryl ring (Figure 7(b) and (d)), which is lost only upon addition to PTI – a process that incurs an energy penalty.

Optimised structures from the addition of ketenimine (a, c), nitrene (b, d), 4-fluoroketenimine (e, g) and 4-fluoronitrene (f, h) to one -NH bridge per void of the PTI monolayer.
It is important at this point to differentiate the addition of nitrene to PTI discussed here to the functionalisation of graphene with the same molecule. In both cases, the product formed contains the aromatic ring in a direction normal to the 2D layer; however, in the case of graphene, this geometry is due to formation of an aziridine through addition of the nitrene radical to a C=C double bond, while for PTI the product is the insertion of nitrene in an N–H bond, with the new linkage requiring a bent geometry to avoid steric repulsion from the carbon nitride plane.
The optimised structures resulting from nitrene/ketenimine interaction with the −NH2 groups located at the edges of the PTI ribbon are shown in Figure 8. The same distortion of the seven-membered ring in the as formed azepine species is seen in all models and functionalisation with both ketenimine and nitrene (X = H, F) results in out-of-plane bending to mitigate steric repulsions from the PTI framework.

Optimised structures from the addition of ketenimine (a, c), nitrene (b, d), 4-fluoroketenimine (e, g) and 4-fluoronitrene (f, h) to the -NH2 edge sites of the PTI monolayer.
The reaction energies were more negative for the addition of ketenimine and nitrene to the edges compared to those at the bridge sites of PTI, where there are fewer steric constraints. The more negative reaction energies when functionalising the edges confirm preferential addition to the terminal –NH2 groups rather than the in-layer –NH bridges as was already discussed for the methylation reaction. The fluorination of the aryl ring in 1-azido-4-fluorobenzene, generating 4-fluoronitrene and 4-fluoroketenimine upon thermal degradation, shifted the reaction energies by a small amount that did not significantly modify the trends as shown in Table 3.
The fraction of reacted edges in the cleaved monolayer was changed in increments between 1/4 and 1 to examine the effect of surface coverage. The edge-rich model was functionalised from either a single side or both sides, and with all or alternating edge sites modified. As presented in Table 3, the reaction energies for the nitrene addition remain largely unchanged regardless of the degree of surface coverage, between −1.85 and −1.87 eV for the H substituted reactant, and −1.83 and −1.86 eV for the halogenated species. However, for the ketenimine product, we observe a larger variation in the energies. The reaction energies are more negative when both sides of the ribbon are functionalised. For non-halogenated reactants the energy decreases from −1.58 to −1.97 eV, and for the fluorinated species from −1.66 to −2.07 eV, indicating that a higher surface coverage of ketenimine is favourable. The cooperative effects resulting from higher coverage could potentially release residual strain arising at the lower level of functionalisation.
Organo-radical addition: experimental results
We then tested the addition of azidobenzene and 1-azido-4-fluorobenzene to PTI in DMSO at 90 °C experimentally, with the expectation that ketenimine (X = H, F) would react with predominantly the −NH2 edge sites of PTI and decorate the surface with azepine species. No new peaks appear in the FTIR spectra (Figure 9(a)) around wavenumber 3000 cm−1 that is associated with the unsaturated =C–H stretch in alkenes and aromatic compounds, of which should have been present had the reaction of either azidobenzene or 1-azido-4-fluorobenzene with PTI proved successful. 29 In fact, the only spectral change noted was the peak emerging at wavenumber 1016 cm−1, that is most likely due to some modification of the PTI network caused by prolonged heating in solvent, as the same peak appears in the control experiment. X-ray photoelectron spectroscopy (XPS) measurements (Figure 9(c)) detected no fluorine components present in the PTI phase after the attempted reaction with 1-azido-4-fluorobenzene, indicating that the reaction between ketenimine and the amine sites of PTI was unsuccessful. The maximum binding energy derived from the C1s and N1s scans shifted to higher binding energies (289 to 290 eV and 399 to 401 eV, respectively) upon functionalisation, indicating a decrease in the surrounding electron density, most likely due to introduced damage or disorder. Some broadening of the PTI peaks along with the appearance of broad background features was apparent in the XRD patterns of the samples following functionalisation attempts, indicating that there were some effects on the crystallinity. The (100) reflection associated with the in-plane separation of the triazine rings decreased in intensity following the attempted functionalisation reaction.14,30
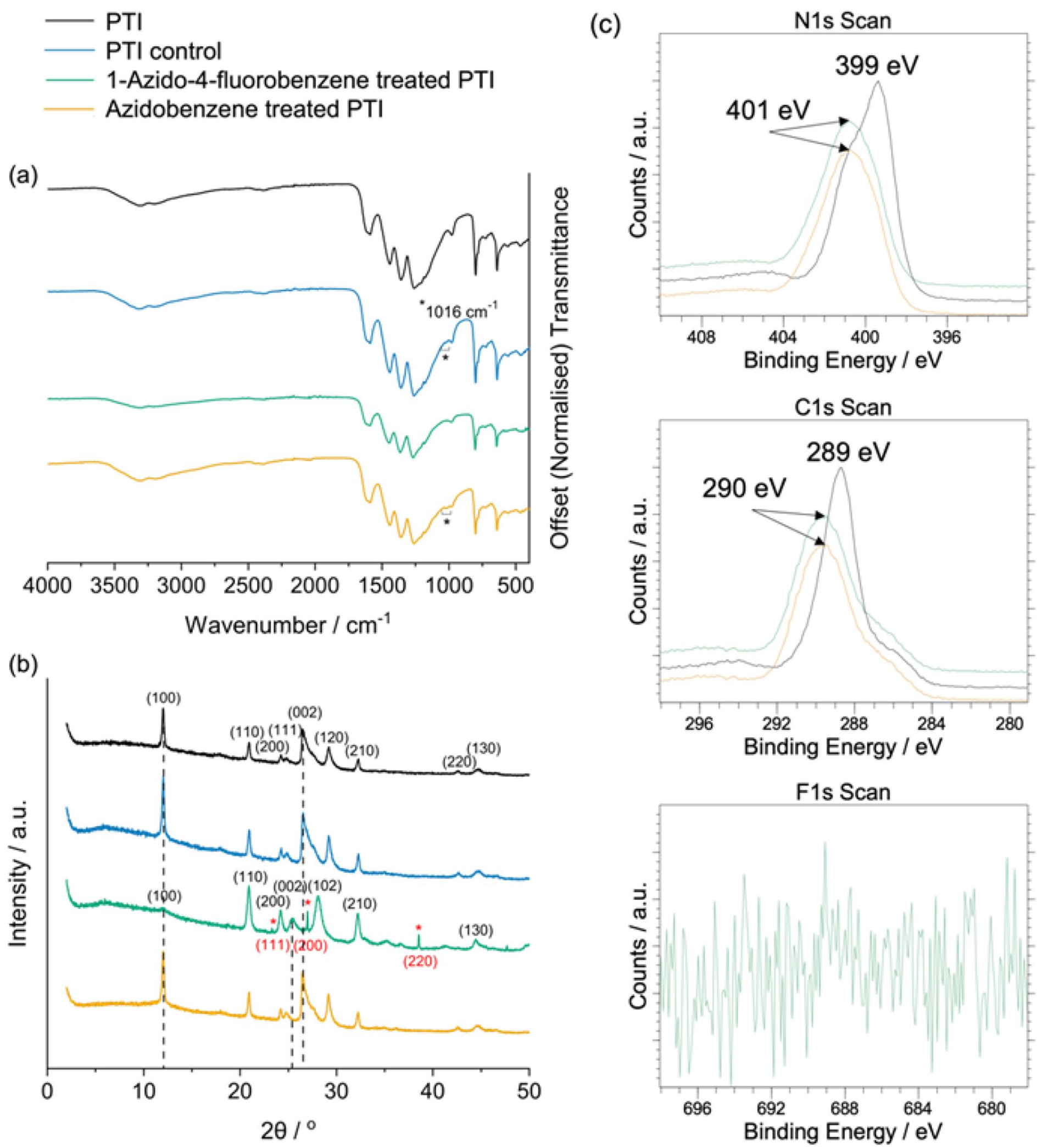
(a) FTIR spectra, (b) XRD patterns and (c) XPS scans of the PTI starting material, the control experiment and PTI treated with azidobenzene and 1-azido-4-fluorobenzene.
Conclusion
We have investigated functionalisation reactions of crystalline layered carbon nitride materials with the PTI structure. DFT results showed that functionalisation with alkyl species such as methyl using alkyl halide reagents is unlikely to be successful, and indeed, no changes in the sample were observed to occur in experimental trials. Functionalisation occurred preferentially at the side surfaces of PTI that are present in lower concentration, impacting the extent to which organic additions will change the overall solubility of PTI in solvent. Furthermore, since PL depends mostly on layer stacking, it is unlikely to be heavily affected by functionalising the −NH2 edges of PTI. However, the DFT calculations indicated that addition of reactive nitrene and ketenimine intermediates, generated from decomposition of azidobenzene, to the N–H bonds of PTI is thermodynamically favourable, although it relies on the formation of high energy intermediates and is not likely to be feasible via thermolysis routes. This was borne out by the negative results of the experimental trials that might not have produced nitrene or ketenimine species. Future work should focus instead on photoinitiation of azidobenzene degradation to result in successful functionalisation of PTI.14,30
Methodology
Computational details
The structures of the reactants and products for the functionalisation reactions were optimised using Density Functional Theory (DFT), as implemented in the CRYSTAL17 code. 31 The calculations were carried out using the B3LYP hybrid exchange functional, augmented with the D3 semi-empirical dispersion correction to account for dispersion forces.32,33 The D3 term was evaluated as a sum of two- and three-body contributions to the dispersion energy. The geometrical counterpoise scheme was used to remove artificial overbinding effects arising from the Basis Set Superposition Error (BSSE).34,35 For the electronic description of the atomic orbitals Gaussian basis sets available from the CRYSTAL online database were selected and are indicated by the following labels: H_3-1p1G_gatti_1994, C_6-31d1G_gatti_1994, N_6-31d1G_gatti_1994, O_6-31d1_gatti_1994, I_HAYWLC-31G_prencipe_1990, and F_7-311G_nada_1993.36,37 To sample reciprocal space, the Monkhorst net was defined with a shrinking factor equal to 6 along all lattice vectors. 38 Default geometry optimisation criteria were used, with TOLDEE threshold set to 7 and TOLINTEG truncation criteria set to 7, 7, 7, 7, 14. We used spin polarised calculations to determine the energy of the nitrene diradical in a triplet state. PTI-methyl out-of-plane displacements were calculated using the difference in z-coordinate between the average z-coordinate of PTI constituent atoms (C, N, H) and the methyl carbon; methyl hydrogens were discounted. PTI-methyl out-of-plane angles were calculated from an average of torsional angles of N-C-N-CMethyl bond motifs, as the macroscopic PTI plane deformed after relaxation. Each bridge methyl had four angles measured (Na-Cx-N-CMethyl; Nb-Cx-N-CMethyl; Nc-Cy-N-CM; Nd-Cy-N-CMethyl), while each edge site had two torsional anglers averaged (Na-Cx-N-CMethyl; Nb-Cx-N-CMethyl).
Preparation of PTI
PTI-LiBr was prepared as described by Miller et al. 13 In short, DCDA was condensed in a molten LiBr/KBr flux to yield LiBr-intercalated PTI which was fully deintercalated using Soxhlet extraction, with deionised water over 2 days, before heating at 100 °C under vacuum (~10 mbar) for 1 h in a sealable tube to yield crystalline intercalant-free PTI with a layer composition C6N9H3. The resultant intercalant-free PTI was sealed and stored in an N2 glovebox.
Methylation of PTI
Intercalant-free PTI (17.0 ± 0.1 mg) with potassium carbonate (K2CO3: (1.0074 ± 0.0001 g) in DMAc (20 ± 0.5 mL) was stirred in a Suba-Sealed round bottom flask, under N2. CH3I (1.000 ± 0.005 mL, 16 mmol, ~60 eq vs N–H in PTI) was added slowly to prevent overheating, and the reaction mixture stirred at room temperature for 24 h. The resulting product was filtered under vacuum using a mixed cellulose 220 nm membrane and rinsed with deionised water three times to dissolve any remaining K2CO3. The product was dried under vacuum for 24 h to yield a brown powder (0.0129 ± 0.0001 g, 76%).
Organo-radical addition of PTI
Intercalant-free PTI (80.0 ± 0.1 mg) was sealed and suspended in DMSO (40.0 ± 0.5 mL) and sonicated in the RS Pro Digital Ultrasonic Cleaner for 2 h at 100 W. The resulting suspension of PTI (2 mg mL−1, 35.0 ± 0.5 mL) was diluted with additional DMSO (25.0 ± 0.5 mL) and stirred in a Suba-Sealed three-neck round-bottom flask attached to an air condenser. A 0.5 M tert-butyl methyl ether solution of either azidobenzene or 1-azido-4-fluorobenzene (0.03 M, 3.600 ± 0.005 mL) was added slowly and the reaction mixture was purged with N2 for 15 min. An N2 balloon was fitted to the round-bottom flask and the reaction mixture was heated at 90 °C for 20 h. The resulting product was filtered under vacuum using a PTFE 0.1 μm membrane and rinsed with acetone. The product was dried under vacuum to yield a brown powder. A control experiment was carried out following the same procedure using PTI (0.0800 ± 0.0001 g) alone suspended in DMSO (40.0 ± 0.5 mL), that is, without addition of the azide.
Footnotes
Acknowledgements
The authors acknowledge the use of the UCL Kathleen High Performance Computing Facility (Kathleen@UCL), and associated support services, in the completion of this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this paper.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: AJC would like to thank the Society of Chemical Industry and the Ramsay Memorial Trust for funding. This project was partly supported by funding from the EU Graphene Flagship under Horizon 2020 Research and Innovation programme grant agreement No. 785219-881603-GrapheneCore3. Via our membership of the UK’s HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/R029431), this work used the ARCHER2 UK National Supercomputing Service (![]() ) and the UK Materials and Molecular Modelling Hub, which is partially funded by EPSRC (EP/T022213).
) and the UK Materials and Molecular Modelling Hub, which is partially funded by EPSRC (EP/T022213).