Experimental
General directions
Starting materials were obtained from commercial suppliers and were used without further purification unless otherwise stated. CAS number for (S)-4-[(tert-butyldimethylsilyl)oxy]cyclopent-2-en-1-one (3): 61305-36-0. All commercially available solvents were used as supplied unless otherwise stated. All ‘dry’ solvents were dried and distilled by standard procedures. Glassware was either dried in an oven, or flame-dried with a Bunsen burner before use and assembled hot then cooled to room temperature under a stream of nitrogen. Oxygen-free, anhydrous nitrogen was obtained from BOC. Thin-layer chromatography (TLC) was carried out on Merck silica gel aluminium sheets (60 F254). Ultraviolet (UV) light and a mixture of KMnO4 (1.5 g), K2CO3 (10 g), 10% NaOH (1.25 mL) in water (200 mL) was used to visualise spots. Merck silica 60 Å (230–400 mesh) 9385 was used for flash column chromatography. Nuclear magnetic resonance (NMR) spectra were recorded on Varian Unit 300, 400 or 500 MHz spectrometers as specified. Spectra were calibrated using trimethylsilane (TMS) or the residual protiated solvent. Coupling constants (J) are quoted in Hertz. 1H and 13C NMR chemical shift assignments are based on two-dimensional NMR techniques, including 1H-1H-gCOSY and heteronuclear single quantum coherence (HSQC) experiments. All values are reported in parts per million (ppm). Infrared (IR) spectra were recorded on a Bruker Alpha Fourier-transform infrared (FTIR) spectrometer. High-resolution mass spectrometry (HRMS) was performed using a Waters Crop, Micromass LCT, electrospray ionisation (ESI) spectrometer. Melting points were determined in an open capillary on a Gallenkamp melting point apparatus and are uncorrected. Compound names were generated using ChemDraw software. Known compounds are referenced accordingly.
(1R,4S)-4-[(tert-Butyldimethylsilyl)oxy]cyclopent-2-enol (4a) and (1S,4S)-4-[(tert-butyldimethylsilyl)oxy]cyclopent-2-enol (4b)
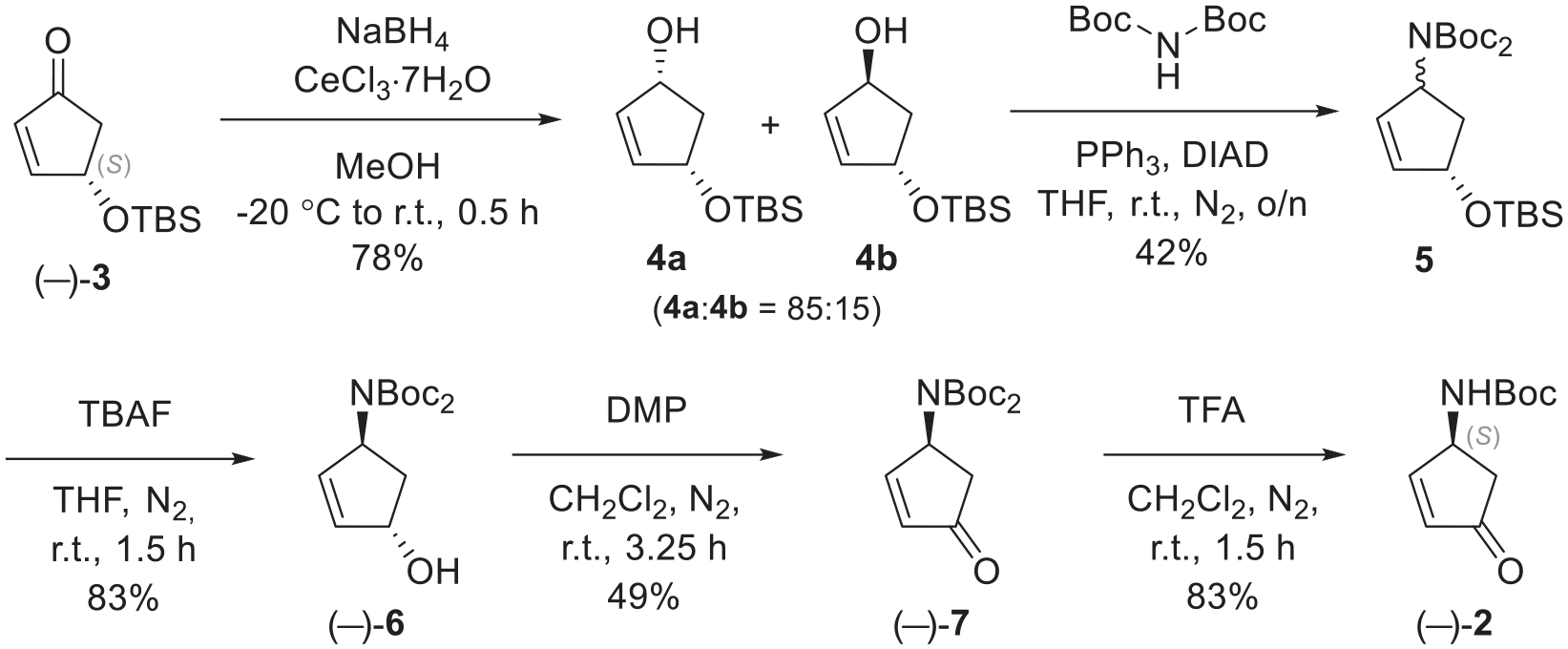
Cerium chloride heptahydrate (1.76 g, 4.72 mmol, 1.0 equiv.) was added to a solution of (S)-4-(tert-butyldimethylsilyl)cyclopent-2-enone (3) (1.01 g, 4.76 mmol, 1.0 equiv.) in MeOH (15 mL) and the resulting suspension was stirred vigorously for 5 min until fully dissolved.
23
The solution was cooled to −20 °C (adjusted with dry ice in acetone) and sodium borohydride (0.18 g, 4.76 mmol, 1.0 equiv.) was added slowly. After stirring at this temperature for 10 min, the cooling bath was removed and the suspension was stirred at room temperature for an additional 20 min. Following this, sat. NH4Cl (40 mL) was added dropwise and the mixture was then extracted with CH2Cl2 (3 × 50 mL). The combined organic phases were dried over MgSO4, filtered and the solvent removed in vacuo, which resulted in the formation of the desired product 4 (0.79 g, 78%) as a colourless oil and as a mixture of diastereomers. The crude mixture, containing 15% of the diastereomer 4b (as determined by 1H NMR spectroscopy), with data as reported,
23
was used further without additional purification. Data for the major diastereomer 4a: 1H NMR (400 MHz, CDCl3): δ 0.09 (s, 6H, CH3), 0.90 (s, 9H, CH3), 1.52 (dt, J = 14.0, 4.5 Hz, 1H, CH2), 1.85 (br s, 1H, OH), 2.69 (dt, J = 14.0, 7.0 Hz, 1H, CH2), 4.59 (br s, 1H, CH), 4.64–4.69 (m, 1H, CH), 5.89 (d, J = 5.5 Hz, 1H, CH), 5.95 (d, J = 5.5 Hz, 1H, CH) ppm;13C NMR (101 MHz, CDCl3): δ −4.55 (CH3), −4.5 (CH3), 18.3 (C), 26.0 (CH3), 44.8 (CH2), 75.3 (CH), 75.35 (CH), 135.8 (CH), 137.1 (CH) ppm (see below for NMR spectroscopic data for 4b).
Di-tert-butyl{(1S,4S)-4-[(tert-butyldimethylsilyl)oxy]cyclopent-2-en-1-yl}imidodicarboxylate [(–)-5]:
Alcohol 4 (0.36 g, 1.68 mmol, 1.0 equiv.) was added to a solution of triphenylphosphine (1.32 g, 5.03 mmol, 3.0 equiv.) and di-tert-butyl-iminodicarboxylate (0.55 g, 2.53 mmol, 1.5 equiv.) in dry THF (20 mL). Diisopropyl azodicarboxylate (0.82 mL, 4.16 mmol, 2.5 equiv.) was added, and the solution was stirred at room temperature for 18 h. After this time, the reaction mixture was concentrated in vacuo and purified by column chromatography (c-Hex/EtOAc; 95:5→9:1), affording the desired product (−)-5 (0.295 g, 42%) as a colourless oil. Rf = 0.5 (c-Hex/EtOAc; 9:1); IR (neat): νmax = 3007, 2979, 2955, 2857, 1743, 1704, 1473, 1461, 1391, 1346, 1252, 1149, 1113 cm−1; 1H NMR (400 MHz, CDCl3): δ 0.07 (br s, 6H, CH3), 0.89 (s, 9H, CH3), 1.48 (s, 18H, CH3), 2.06 (ddd, J = 14.0, 8.5, 2.5 Hz, 1H, CH2), 2.24 (ddd, J = 14.0, 7.5, 5.0 Hz, 1H, CH2), 5.03–5.09 (m, 1H, CH), 5.41–5.47 (m, 1H, CH), 5.78–5.85 (m, 2H, CH) ppm; 13C NMR (101 MHz, CDCl3): δ −4.5 (CH3), −4.45 (CH3), 18.4 (C), 26.1 (CH3), 28.1 (CH3), 39.5 (CH2), 62.0 (CH), 77.2 (CH), 82.4 (C), 133.8 (CH), 135.8 (CH), 153.1 (CO) ppm; HRMS (ES+): m/z C21H39NO5SiNa (MNa+) calcd. 436.2490; found 436.2493; [α]D = −112.6 (c = 0.1, CHCl3).
Di-tert-butyl[(1S,4S)-4-hydroxycyclopent-2-en-1-yl]imidodicarboxylate [(−)-6]
A 1 M solution of TBAF in THF (3.02 mL, 3.02 mmol, 1.5 equiv.) was added to a solution of TBS ether 5 (831 mg, 2.01 mmol, 1.0 equiv.) in dry THF (30 mL). The solution was stirred at room temperature for 1 h 30 min. After this time, the reaction mixture was concentrated under a flow of air and the resulting residue was directly purified by column chromatography (c-Hex/EtOAc; 2:1), affording the desired product (−)-6 (498 mg, 83%) as a white solid. M.p. = 60–62 °C; Rf = 0.5 (c-Hex/EtOAc; 1:1); IR (neat): νmax = 3250, 2980, 2933, 1746, 1706, 1477, 1456, 1390, 1345 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.49 (s, 18 H, CH3), 2.11 (ddd, J = 14.0, 8.5, 2.0 Hz, 1H, CH2), 2.34 (ddd, J = 14.0, 7.5, 5.5 Hz, 1H, CH2), 5.06 (br s, 1H, CH), 5.44–5.53 (m, 1H, CH), 5.91 (ddd, J = 5.5, 2.0, 1.0 Hz, 1H, CH), 5.93–5.98 (m, 1H, CH) ppm; 13C NMR (101 MHz, CDCl3): δ 28.1 (CH3), 39.3 (CH2), 61.8 (CH), 76.8 (CH), 82.6 (C), 134.8 (CH), 135.6 (CH), 153.0 (CO) ppm; HRMS (ES+): m/z C15H26NO5 (MH+) calcd. 300.1805; found 300.1806; [α]D = −142.6 (c = 0.1, CHCl3).
Di-tert-butyl[(1S)-4-oxocyclopent-2-en-1-yl]imidodicarboxylate [(−)-7]
Dess–Martin periodinane (651 mg, 1.53 mmol, 1.0 equiv.) was added to a solution of alcohol 6 (441 mg, 1.47 mmol, 1.0 equiv.) in dry CH2Cl2 (40 mL). The solution was stirred at room temperature for 2.5 h. (The reaction was monitored by TLC.) An additional aliquot of Dess–Martin periodinane (126 mg, 0.30 mmol, 0.2 equiv.) was added, and the reaction mixture was stirred for a further 45 min. The reaction mixture was filtered through Celite and concentrated under a flow of air. The resulting residue was purified by column chromatography (c-Hex/EtOAc; 4:1), affording the desired product (−)-7 (216 mg, 49%) as a colourless solid. M.p. = 62–64 °C; Rf = 0.3 (c-Hex/EtOAc; 4:1); IR (neat): νmax = 2976, 2925, 2852, 1750, 1705, 1367, 1345, 1262, 1232, 1134, 1101 cm−1; 1H NMR (500 MHz, CDCl3): δ 1.49 (s, 18 H, CH3), 2.54 (dd, J = 18.0, 3.0 Hz, 1H, CH2), 2.73 (dd, J = 18.0, 7.0 Hz, 1H, CH2), 5.48–5.53 (m, 1H, CH), 6.22 (dd, J = 5.5, 2.5 Hz, 1H, CH), 7.52 (dd, J = 5.5, 2.5 Hz, 1H, CH) ppm; 13C NMR (101 MHz, CDCl3): 28.1 (CH3), 40.6 (CH2), 56.1 (CH), 83.7 (C), 134.1 (CH), 152.3 (CH), 163.0 (CO), 206.3 (CO) ppm; HRMS (ES+): m/z C15H23NO5Na (MNa+) calcd. 320.1468; found 320.1471; [α]D = −82.4 (c = 0.1, CHCl3).
(S)-tert-Butyl (4-oxocyclopent-2-en-1-yl)carbamate [(−)-2]
Trifluoroacetic acid (70 μL, 0.97 mmol, 1.9 equiv.) was added to a solution of cyclopentenone (−)-7 (152 mg, 0.51 mmol, 1.0 equiv.) in dry CH2Cl2 (7 mL) and the solution was stirred under an inert atmosphere for 1.5 h. After this time, the solution was diluted with CH2Cl2 (8 mL) and washed with NaHCO3 (10 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL) and the combined organic phases were dried over MgSO4 and the solvent removed in vacuo. Purification by column chromatography (c-Hex/EtOAc; 2:1) afforded the desired product (−)-2 (83 mg, 83%) with data as reported.11–15 M.p. = 110–111 °C; Rf = 0.2 (c-Hex/EtOAc; 2:1); IR (neat): νmax = 3325, 2981, 2932, 1719, 1673, 1529, 1366, 1252, 1159 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.46 (s, 9H, CH3), 2.16 (dd, J = 18.5, 2.5 Hz, 1H, CH2), 2.86 (dd, J = 18.5, 6.5 Hz, 1H, CH2), 4.73 (br s, 1H, NH), 4.95 (br s, 1H, CH), 6.24 (dd, J = 5.5, 2.0 Hz, 1H, CH), 7.53 (dd, J = 5.5, 2.5 Hz, 1H, CH) ppm; 13C NMR (101 MHz, CDCl3): 28.5 (CH3), 42.6 (CH2), 51.2 (CH), 80.4 (C), 135.4 (CH), 155.2 (CH), 162.3 (CO), 206.6 (CO) ppm; HRMS (ES+): m/z C10H15NO3Na (MNa+) calcd. 220.0944; found 220.0945; [α]D = −75.5 (c = 0.1, CHCl3), {Lit.
11
[α]D = −73 (c = 0.1, CHCl3)}.
(1S,4S)-4-((tert-Butyldimethylsilyl)oxy)cyclopent-2-enol (4b)
Alcohol 4 (100 mg, 0.47 mmol, 1.0 equiv.) was added to a solution of triphenylphosphine (241 mg, 0.92 mmol, 2.0 equiv.) and benzoic acid (88 mg, 0.71 mmol, 1.5 equiv.) in dry THF (5 mL). The solution was cooled to 0 °C and DIAD (0.2 mL, 0.94 mmol, 2.0 equiv.) was added. Following this, the reaction was allowed to warm to room temperature and stirred for 16 h under an inert atmosphere. After this time, the solvent was removed under a flow of air and the residue was diluted with Et2O (10 mL) and washed with NaHCO3 (3 × 10 mL). The organic phase was dried over MgSO4 and the solvent removed in vacuo. The intermediate was then diluted in methanol (5 mL) and K2CO3 (326 mg, 2.35 mmol, 5.0 equiv.) was added. The resulting suspension was stirred under an inert atmosphere for 4 h, after which the suspension was filtered and concentrated in vacuo. The resulting residue was dissolved in Et2O (10 mL) and water (10 mL) and washed with water (3 × 10 mL) and brine (10 mL). The organic phase was dried over MgSO4 and the solvent removed in vacuo. Purification by column chromatography (c-Hex/EtOAc; 3:1) afforded the desired product 4b (12 mg, 12%) with data as reported.
23
1
H NMR (400 MHz, CDCl3): δ 0.06−0.10 (m, 6H, CH3), 0.89 (s, 9H, CH3), 1.96−2.10 (m, 2H, CH2), 5.02 (s, 1H, CH), 5.06−5.12 (m, 1H, CH), 5.91−5.97 (m, 2H, CH) ppm; 13C NMR (101 MHz, CDCl3): −4.5 (CH3), 18.4 (C), 26.1 (CH3), 44.7 (CH2), 76.4 (CH), 76.7 (CH), 135.6 (CH), 138.6 (CH) ppm.
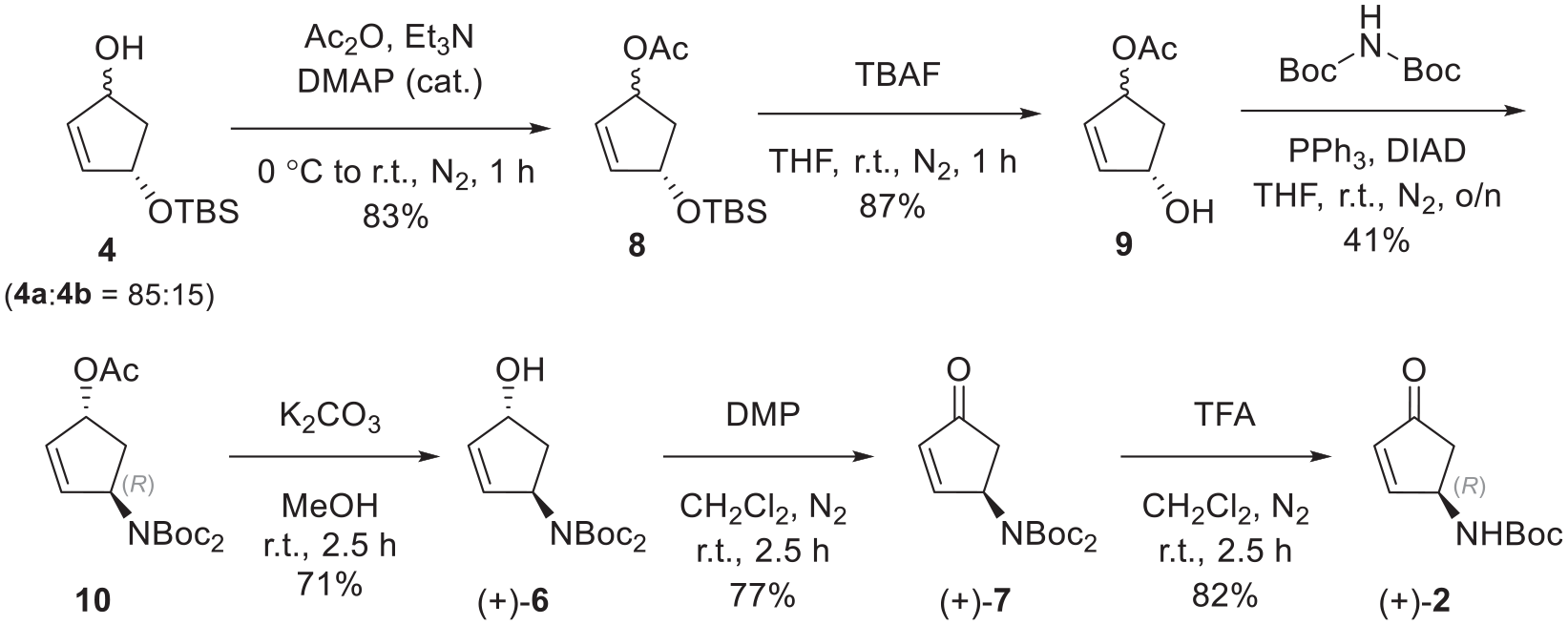
(1R,4S)-4-((tert-Butyldimethylsilyl)oxy)cyclopent-2-en-1-yl acetate (8)
DMAP (cat.) was added to a solution of alcohol 4 (785 mg, 3.66 mmol, 1.0 equiv.) in dry CH2Cl2 (30 mL). The solution was cooled to 0 °C and Et3N (0.70 mL, 5.02 mmol, 1.3 equiv.) was added, followed by acetic anhydride (0.40 mL, 4.23 mmol, 1.2 equiv.). The cooling bath was removed, and the solution was stirred for 2.5 h. After this time, NH4Cl (20 mL) was added to the solution and the resulting mixture was stirred for 10 min. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (2 × 20 mL). The combined organic phase was washed with NaHCO3 (40 mL) and dried over MgSO4. Filtration and solvent removal in vacuo gave the desired product 8 (782 mg, 3.05 mmol, 83%) as a yellow liquid. The crude product (8), with data as reported,
26
was used without further purification. 1H NMR (400 MHz, CDCl3): δ 0.04–0.12 (m, 6H, CH3), 0.90 (s, 9H, CH3), 1.61 (dt, J = 14.0, 5.0 Hz, 1H, CH2), 2.05 (s, 3H, CH3), 2.81 (dt, J = 14.0, 7.5 Hz, 1H, CH2), 4.68–4.76 (m, 1H, CH), 5.42–5.50 (m, 1H, CH), 5.89 (dt, J = 5.5, 1.5 Hz, 1H, CH), 5.97 (dt, J = 5.5, 1.5 Hz, 1H, CH) ppm; 13C NMR (101 MHz, CDCl3): −4.54 (CH3), −4.49 (CH3), 18.3 (C), 21.3 (CH3), 26.0 (CH3), 41.3 (CH2), 75.0 (CH), 77.1 (CH), 131.3 (CH), 139.1 (CH), 171.0 (CO) ppm.
(1R,4S)-4-Hydroxycyclopent-2-en-1-yl acetate (9)
A 1 M solution of TBAF in THF (2.8 mL, 2.80 mmol, 1.15 equiv.) was added to a solution of TBS ether 8 (637 mg, 2.48 mmol, 1.0 equiv.) in dry THF (35 mL). The solution was stirred at room temperature for 1 h. After this time, the reaction mixture was concentrated under a flow of air, and the resulting residue was purified by column chromatography (c-Hex/EtOAc; 1:1), affording the desired product 9 (308 mg, 87%) as a white solid, with data as reported.
26
1
H NMR (400 MHz, CDCl3): δ 1.65 (dt, J = 14.5, 4.0 Hz, 1H, CH2), 2.05 (s, 3H, CH3), 2.81 (dt, J = 14.5, 7.5 Hz, 1H, CH2), 4.69–4.75 (m, 1H, CH), 5.46–5.53 (m, 1H, CH), 5.98 (ddd, J = 5.5, 2.0, 1.0 Hz, 1H, CH), 6.08–6.15 (m, 1H, CH) ppm; 13C NMR (101 MHz, CDCl3): 21.3 (CH3), 40.6 (CH2), 74.9 (CH), 77.2 (CH), 132.6 (CH), 138.7 (CH), 171.0 (CO) ppm.
Di-tert-butyl[(1S,4R)-4-acetoxycyclopent-2-en-1-yl]imidodicarboxylate (10)
Alcohol 9 (0.29 g, 2.04 mmol, 1.0 equiv.) was added to a solution of triphenylphosphine (1.59 g, 6.05 mmol, 3.0 equiv.) and di-tert-butyl-iminodicarboxylate (0.67 g, 3.07 mmol, 1.5 equiv.) in dry THF (30 mL). Diisopropyl azodicarboxylate (1.00 mL, 5.08 mmol, 2.5 equiv.) was added and the solution was stirred at room temperature for 18 h. After this time, the reaction mixture was concentrated and purified by column chromatography (c-Hex/EtOAc; 9:1), affording the desired product 10 (0.287 g, 41%) as a colourless oil. Rf = 0.4 (c-Hex/EtOAc; 9:1); IR (neat): νmax = 2980, 2936, 1736, 1701, 1344, 1234, 1146, 1112 cm−1; 1H NMR (400 MHz, CDCl3): δ 1.48 (s, 18 H, CH3), 2.02 (s, 3H, CH3), 2.19 (ddd, J = 14.5, 8.0, 2.0 Hz, 1H, CH2), 2.35 (ddd, J = 14.5, 7.5, 5.5 Hz, 1H, CH2), 5.41–5.51 (m, 1H, CH), 5.77–5.82 (m, 1H, CH), 5.89–5.96 (m, 1H, CH), 6.02 (dd, J = 5.5, 2.0 Hz, 1H, CH) ppm; 13C NMR (101 MHz, CDCl3): 21.3 (CH3), 28.1 (CH3), 36.0 (CH2), 61.4 (CH), 79.4 (CH), 82.7 (C), 130.5 (CH), 138.3 (CH), 152.8 (CO), 171.1 (CO) ppm; HRMS (ES+): m/z C17H27NO6Na (MNa+) calcd. 364.1731; found 364.1732; [α]D = +150.4 (c = 0.1, CHCl3).
Di-tert-butyl[(1R,4R)-4-hydroxycyclopent-2-en-1-yl]imidodicarboxylate [(+)-6]
K2CO3 (172 mg, 1.24 mmol, 2.0 equiv.) was added to a solution of acetate 10 (210 mg, 0.62 mmol, 1.0 equiv.) in MeOH (5 mL). The solution was stirred for 2 h, after which NaHCO3 (15 mL) was added. The phases were separated, and the aqueous phase was extracted with EtOAc (3 × 15 mL). The combined organic phases were dried over MgSO4 and the solvent removed in vacuo. Purification by column chromatography (c-Hex/EtOAc; 3:1), afforded the desired product (+)-6 (134 mg, 71%) as a colourless waxy solid with data as described above for (−)-6. Rf = 0.2 (c-Hex/EtOAc; 3:1); [α]D = +142.6 (c = 0.1, CHCl3).
Di-tert-butyl[(1S)-4-oxocyclopent-2-en-1-yl]imidodicarboxylate [(+)-7]
Dess–Martin periodinane (561 mg, 1.32 mmol, 1.2 equiv.) was added to a solution of alcohol (+)-6 (328 mg, 1.10 mmol, 1.0 equiv.) in dry CH2Cl2 (30 mL) and the solution was stirred at room temperature for 2.5 h. Following this, the reaction mixture was filtered through Celite, concentrated in vacuo and the resulting residue was purified by column chromatography (c-Hex/EtOAc; 4:1), affording the desired product (+)-7 (251 mg, 77%) as a colourless solid with data as previously stated for (−)-7. [α]D = +79.3 (c = 0.1, CHCl3).
(R)-tert-Butyl (4-oxocyclopent-2-en-1-yl)carbamate [(+)-2]:
Trifluoroacetic acid (0.10 mL, 1.31 mmol, 1.9 equiv.) was added to a solution of cyclopentenone (+)-7 (206 mg, 0.69 mmol, 1.0 equiv.) in dry CH2Cl2 (10 mL), and the solution was stirred under an inert atmosphere for 2.5 h. After this time, the solution was diluted with CH2Cl2 (10 mL) and washed with NaHCO3 (15 mL). The aqueous phase was extracted with CH2Cl2 (3 × 15 mL) and the combined organic phases were dried over MgSO4 and the solvent removed in vacuo. Purification by column chromatography (c-Hex/EtOAc; 2:1) afforded the desired product (+)-2 (111 mg, 82%) with data as described above for (−)-2. [α]D = +74.4 (c = 0.1, CHCl3), {Lit.
13
[α]D = +66.8 (c = 1.0, CHCl3)}.