
Abstract
The standard enthalpies of hydrogenation of 29 unsaturated hydrocarbon compounds were calculated in the gas phase by M06-2X theory with the 6-31g(d) and cc-pVXZ, where X = DZ, TZ, QZ, as well as by complete basis set extrapolated level. Geometries of compounds were optimized at the M06-2X/6-31g(d) level. These M06-2X geometries were used in the M06-2X, and extrapolation calculations with cc-pVXZ basis sets. Comparison of calculation and experimental results shows that the mean absolute deviations between the calculated and experimental enthalpies of hydrogenation range from 25.1 to 5.1 kJ mol−1 at M06-2X calculations, and when using cc-pV(DT)Z extrapolated level, the mean absolute deviations have decreased to 2.7. The results of some calculations showed that the deviations from experimental values are located inside the “chemical accuracy” (±1 kcal mol−1≈±4.2 kJ mol−1). Very good linear correlations between experimental and calculated enthalpies of hydrogenation have been obtained at M06-2X/cc-pVTZ and cc-pV(DT)Z extrapolated levels (standard deviation = 3.2 and 3.4 kJ mol−1, respectively).
Keywords
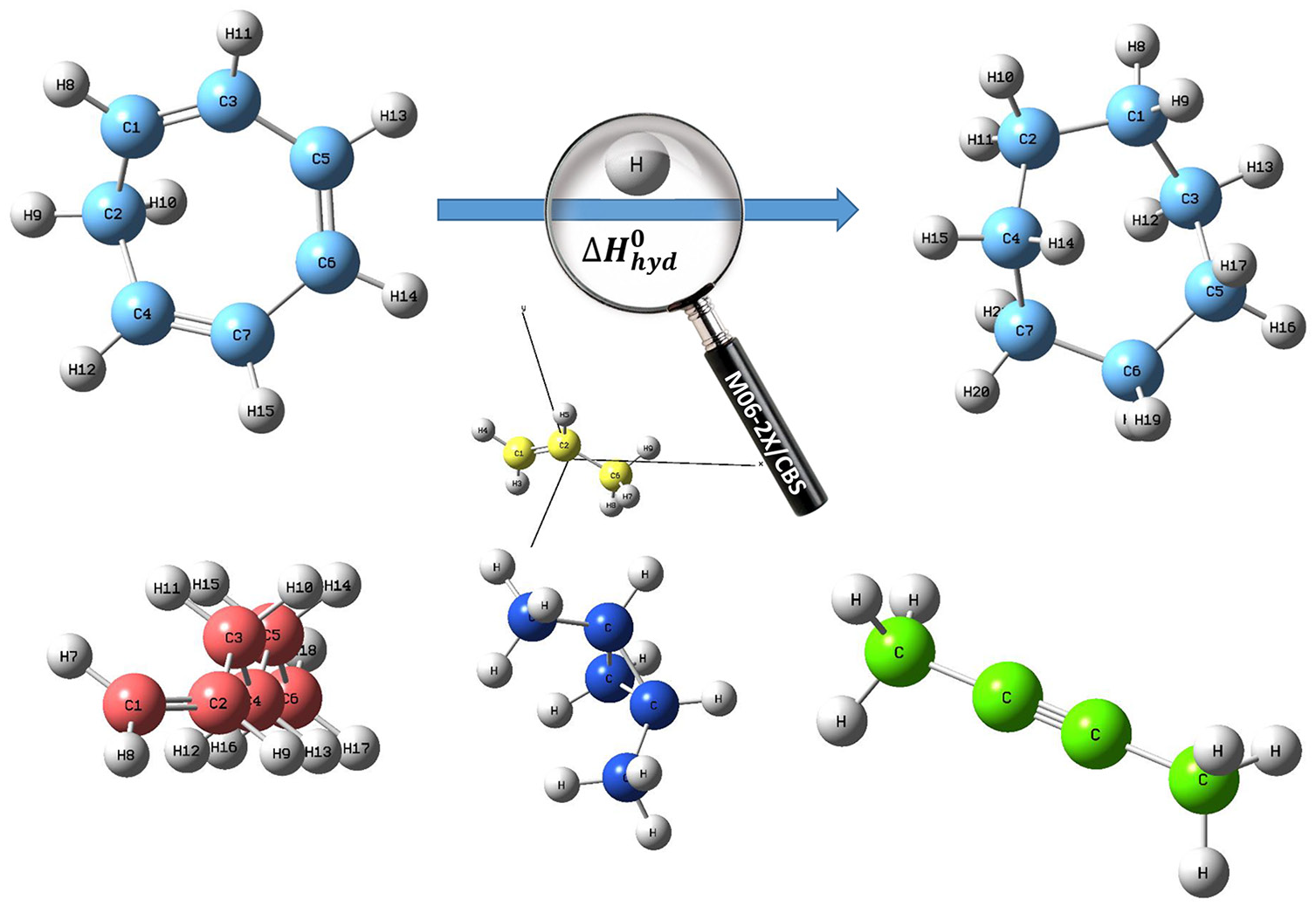
Introduction
Recent advances in computational chemistry have made it possible to calculate enthalpies of formation from quantum-mechanical first principles for rather large unsaturated molecules, some of which are outside the practical range of combustion thermochemistry. Quantum-mechanical calculations of molecular thermochemical properties are, of necessity, approximate. Composite quantum-mechanical procedures may employ approximations at each of several computational steps and may have an empirical factor to correct for the cumulative error. Approximate methods are useful only insofar as the error due to the various approximations is known within narrow limits. Error due to approximation is determined by comparison with a “known” value, but the question of the accuracy of the “known” value immediately arises because the uncertainty of the comparison is determined by the combined uncertainty of the approximate quantum-mechanical result and the standard to which it is compared.
Once the validity of a quantum-mechanical procedure has been established by its ability to reproduce various accurate experimental results, the way is clear to calculate unknown thermochemical values of unstable or explosive compounds, unsuited to classical thermochemical methods, or to calculate thermochemical properties of molecules, radicals, or ions of fleeting existence.1–14 Herein lies a major advantage of the accuracy inherent hydrogen thermochemical results and a reason for renewed interest in the diverse but scattered literature devoted to hydrogen thermochemistry.
The total electronic energy of a molecule is a fundamental quantity in quantum chemistry. By virtue of the Born–Oppenheimer separation of nuclear and electronic motion, this total electronic energy is a function of the nuclear geometric configuration, thereby generating hypersurfaces of potential energy for electronically excited states as well as for the ground state.
It is the purpose of this research to highlight the fact that wavefunction-based quantum-chemical methods can produce molecular electronic energies with an accuracy that rivals or even surpasses that of experimental measurements of molecular energies (in terms of enthalpies of formation).
An important characteristic of the wavefunction-based quantum-chemical methodology is the ability to approach the exact description of the molecular electronic structure in a systematic manner. The ingredients needed to achieve such a systematic approach comprise the use of an advanced hierarchy of wavefunction models, on one hand, and the use of a systematic sequence of basis sets—or a nearly complete basis set (CBS)—of atomic orbitals (AOs), on the other hand.
Basis set convergence
Obviously, the accuracy of computed molecular electronic energies depends on the chosen wavefunction model or density functional. It also depends, however, on the flexibility of the one electron basis set of AOs in terms of which the molecular orbitals (MOs) are expanded.
Of course, basis sets of AOs may be chosen in many different ways (see, for example, the literature15–17). However, for systematic studies of molecular electronic structure, it has proved very useful—at least for wavefunction-based methods—to have a well-defined procedure for generating sequences of basis sets of increasing flexibility. In this manner, hierarchies of basis sets are generated, in which each next higher level represents a systematically improved description of the molecular electronic structure with respect to the next lower level. The basis set hierarchy ends up in an effectively complete basis. 15 This means that, within the hierarchy, the calculated results have converged to within a prescribed accuracy or threshold.
A major problem of wavefunction-based approaches that take into account electron-correlation effects is that the convergence to an effectively complete basis is very slow. To a good approximation, the basis set error is reduced as ∝ N−1 when the number N of AOs in the basis is increased in an optimal manner. Since computing times grow at least as ∝ N4, it is obvious that the computational effort grows much faster than the gain in accuracy. It is therefore no surprise that the accuracy of electron-correlation calculations often is limited by computational, that is, technical constraints.
Correlation-consistent basis sets
The correlation-consistent basis sets of Dunning and co-workers18,19 represent a particularly popular hierarchy of basis sets, in the sense of a principal expansion that can be used to extrapolate to the limit of an effectively complete basis.20–25 If only the valence orbitals are correlated in a calculation, the correlation-consistent polarized valence X-tuple zeta basis sets can be used. These are denoted as cc-pVXZ, where X = D, T, Q, 5, or 6 (double, triple, quadruple, quintuple, or sextuple zeta). This applies to the calculations of this work, where X = D, T and Q. The X in the basis set’s name corresponds to the respective cardinal number (X = 2−6). If all electrons (core as well as valence orbitals) are correlated, correlation-consistent polarized core—valence X-tuple zeta basis sets cc-pCVXZ with 2 ⩽ X ⩽ 5 should be used.
Basis set extrapolation
The basis set hierarchy formed by the correlation-consistent basis sets is well suited for basis set extrapolations of the correlation energy to the limit of a complete set. We can take the limit X→∞ if we know how the correlation energy depends on X.
In 1997, Helgaker et al. 26 introduced the two-point linear extrapolation formula, which is shown in equation (1)
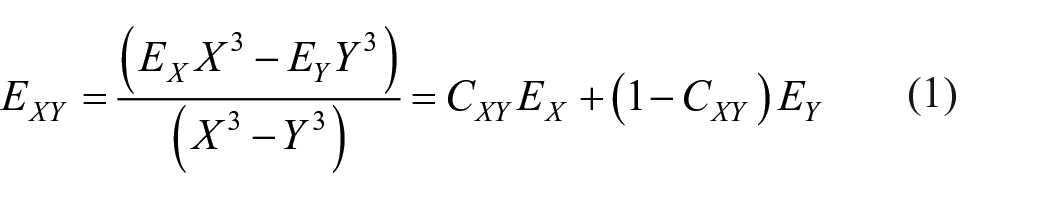
where CXY = X3/(X3 − Y3) is a coefficient, solely determined by the basis sets cc-pCVXZ and cc-pCVYZ used (i.e. not an adjustable parameter), and EX and EY are the electron-correlation energies computed in the basis sets cc-pCVXZ and cc-pCVYZ, respectively.
Equation (1) follows from the observation that for large X, the basis set convergence of electron-correlation effects goes as X−3. This means that the correlation energy EX that is recovered in a given correlation-consistent basis of the type cc-pCVXZ can be expressed as equation (2)

where E∞ is the correlation energy in a complete and infinite basis, and aX−3 represents the leading term of the basis set error. Two equations with two unknowns are obtained if calculations in two different basis sets cc-pCVXZ and cc-pCVYZ are performed. Solving these equations for the two unknowns a and E∞ leads to equation (2), with E∞ taking the value E∞ = EXY.
Experience has shown27–31 that the most accurate estimate of the basis set limit is obtained by extrapolations from two consecutive basis sets with X = Y − 1 (e.g. from the pair cc-pCVTZ/cc-pCVQZ or from cc-pCVQZ/cc-pCV5Z). Results obtained by such extrapolations are referred to as obtained at the cc-pCV(XY)Z level, where XY = DT, TQ, Q5, 56, and so on. Note that only the correlation energy is extrapolated at this level. It is added to the Hartree–Fock energy in the larger of the two basis sets. For example, the total electronic energy at the CCSD(T)(Full)/cc-CV(Q5)Z level consists of the sum of the Hartree–Fock energy in the cc-pCV5Z basis plus the correlation energy extrapolated from the cc-pCVDZ and cc-pCVTZ correlation energies.
In the following section, the M06-2X/cc-pV(XY)Z extrapolation will be applied to obtain estimates of the basis set limit of corrections for M06-2X/6-31g(d) level of theory by calculations of hydrogenation enthalpies of some unsaturated hydrocarbon compounds (Table 1).
Experimental values of hydrogenation enthalpy of some unsaturated hydrocarbons in the gas phase (in kJ mol−1) a .

NIST–JANAF thermochemical tables, Rogers. 32
Enthalpies of hydrogenation
To illustrate the performance of M06-2X calculations in a complete basis, we have computed hydrogenation enthalpies of some unsaturated hydrocarbon compounds as accurately as technically possible. In order to demonstrate the capabilities of the M06-2X approach, we shall compare M06-2X calculations of hydrogenation enthalpies with other quantum-chemical calculations. Furthermore, the examples will illustrate the level of theory and corrections that are needed to obtain accurate molecular energies.
Technical details
Quantum-chemical methods
Electronic energies were computed by the density-functional (M06-2X) 33 approach with cc-pVXZ basis set of Dunning et al., 18 where X = D, T, and Q. The M06-2X/6-31g(d) equilibrium geometries of the reactants and products were optimized with the Gaussian 09 program. 34 All M06-2X calculations were performed at fixed molecular equilibrium geometries that were optimized at the M06-2X/6-31g(d) level. The Cartesian coordinates for all compounds after geometry optimization using M06-2X/6-31g(d) level can be found in supporting information.
Results and discussion
Nonrelativistic electronic energies
The total electronic M06-2X/6-31g(d) energies, Hcorr, Gcorr, and the zero-point vibrational energies (ZPE) are reported in Table 2, while Table 3 shows M06-2X/cc-pVXZ energies computed at the M06-2X/6-31g(d) geometries, where X = D, T, and Q. Table 3 also shows extrapolated values by equation (2) (denoted E∞).
Calculated total electronic energy (Eo), Hcorr, Gcorr, and the zero-point vibrational energies (ZPE) at the M06-2X/6-31g(d) level (in hartree).

Computed M06-2X/ccpVXZ energies at the M06-2X/6-31g(d) geometries, where X = D, T, and Q, as well as extrapolated values by equation (2) (in hartree).

cc-pV(TQ)Z extrapolated level.
cc-pV(DT)Z extrapolated level.
Limit energies were obtained using the web page http://sf.anu.edu.au/~vvv900/cbs/#ref_3. 35
It is noted from Table 3 that the cc-pV(DT)Z extrapolated level yields electronic energies for all reaction components less than cc-pV(QT)Z extrapolated level, and it is expected that the calculations of the hydrogenation enthalpies by cc-pV(DT)Z extrapolated level are compatible with the experimental values.
Enthalpies of hydrogenation at 298.15 K in gas phase
The usual way to calculate enthalpies of reaction is to calculate heats of formation, and take the appropriate sums and difference (equation (3))

However, since the Gaussian program provides the sum of electronic and thermal enthalpies, there is a short cut, namely, to simply take the difference of the sums of these values for the reactants and the products. This works since the number of atoms of each element is the same on both sides of the reaction; therefore, all the atomic information cancels out, and you need only the molecular data. For example, using the information in Table 2 (or Table 3 for energies), the enthalpy of reaction can be calculated simply by equation (4)

E0 can represent either EDZ, ETZ, EQZ or E∞ keeping the calculated Hcorr value at M06-2X/6-31g(d) level is fixed. The calculated enthalpies of hydrogenation are reported in Table 4, along with the experimental values. Table 5 shows statistical parameters for all used computational methods. Figure 1 shows a linear analysis of the best calculated results in terms of experimental results.
Calculated standard enthalpies of hydrogenation of some unsaturated hydrocarbons in the gas phase (in kJ mol−1).

M06-2X/6-31g(d) level.
M06-2X/cc-pVTZ//M06-2X/6-31g(d) level.
M06-2X/cc-pVQZ//M06-2X/6-31g(d) level.
cc-pV(TQ)Z extrapolated level.
cc-pV(DT)Z extrapolated level.
NIST–JANAF thermochemical tables.
Statistical parameters for all used methods to calculate hydrogenation enthalpies (in kJ mol−1).
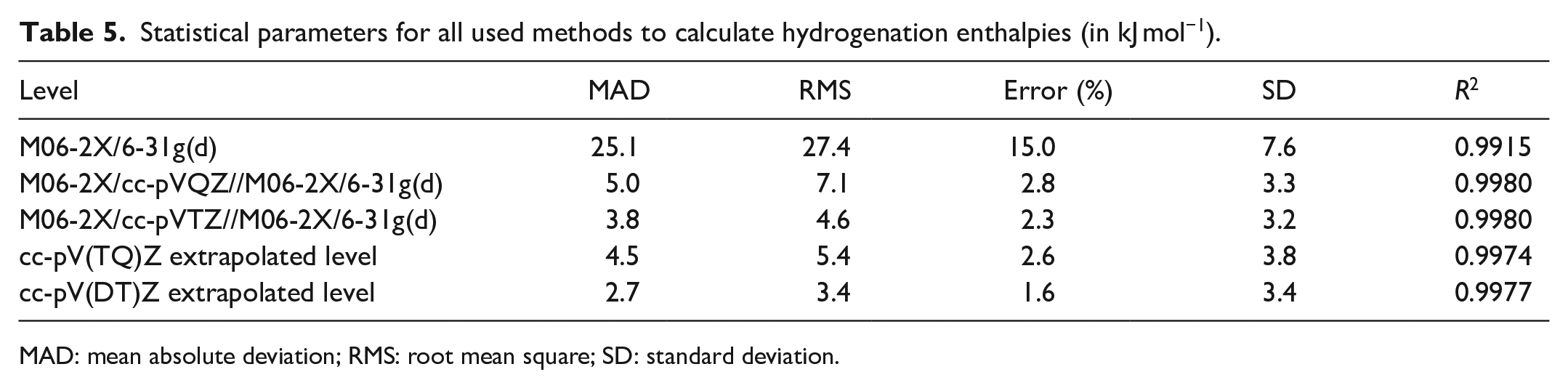
MAD: mean absolute deviation; RMS: root mean square; SD: standard deviation.
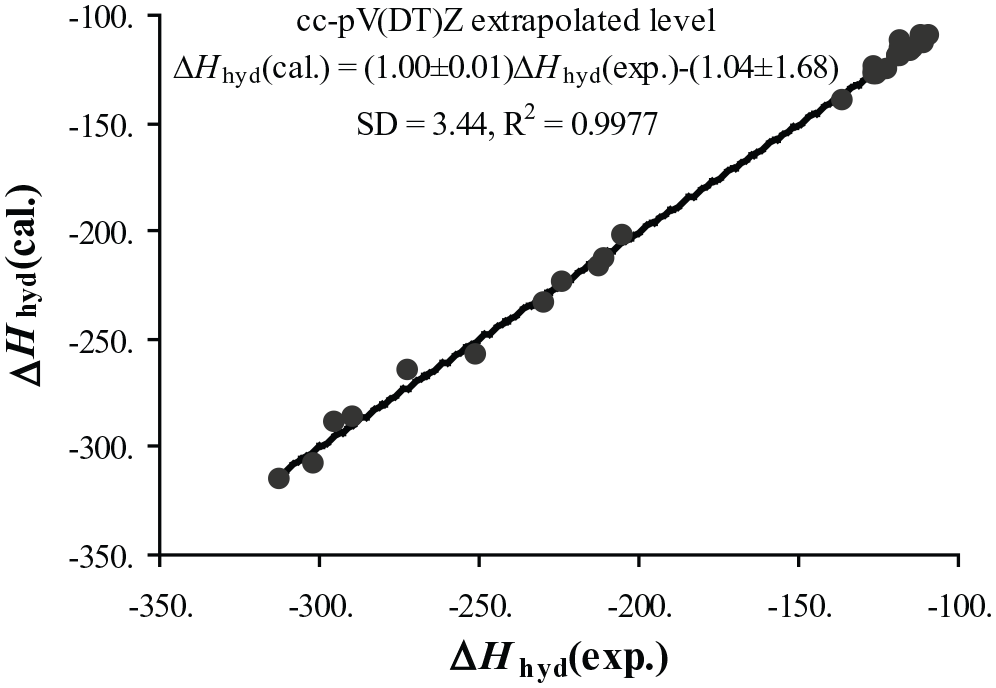
Calculated versus experimental hydrogenation enthalpy
Our best theoretical estimates of the enthalpies of hydrogenation are based on basis set limit extrapolation calculations, when the pair cc-pVDZ and cc-pVTZ are used, and the mean absolute deviation (MAD) between experimental and calculated values is 2.7 kJ mol−1 (Table 5). The enthalpies of hydrogenation of some alkenes (12 compounds) have been calculated at the HF, B3LYP, M06, MP2, G3, G4, CBS-QB3, CBS-APNO, and W1BD levels and, in the case of the first four methods, using a variety of basis sets up to aug-cc-pVTZ, 36 and it is found that the MAD decreases gradually from the first to the last method (12.1–3.2 kJ/mol). Moreover, Rogers and colleagues37–39 calculated the hydrogenation enthalpy for at 298.15 K for reactions involving cyclic and acyclic C4 (20 reactions), cyclic C5 (23 reactions), and C6 (24 reactions) hydrocarbons using the G2 and G2(MP2) ab initio methods, and it is found that the MAD is about 3.3, 3.7, and 5.0 kJ mol−1, respectively.
It is noted from Table 5 that the MAD for the basic set M06-2X/cc-pVQZ level (MAD = 5.0 kJ mol−1) and cc-pV(QT)Z extrapolated level (MAD = 4.5 kJ mol−1) is larger than that for M06-2X/cc-pVTZ level (MAD = 3.8 kJ mol−1) and cc-pV(DT)Z extrapolated level (MAD = 2.7 kJ mol−1) as we expected previously.
Conclusion
Enthalpies of hydrogenation are relatively easy to calculate with density-functional theory (DFT) (M06-2X) giving fairly good agreement with experiment, especially when cc-pVTZ basis sets are used, and basis set extrapolation techniques seem to represent an easy-to-use alternative, especially when the cc-pVDZ and cc-pVTZ basis sets are used.
Supplemental Material
Supporting_Information_All_computed_molecule_Cartesian_coordinates_XYZ – Supplemental material for Calculations of hydrogenation enthalpies of hydrocarbons by M06-2X/CBS extrapolated level in the gas phase
Supplemental material, Supporting_Information_All_computed_molecule_Cartesian_coordinates_XYZ for Calculations of hydrogenation enthalpies of hydrocarbons by M06-2X/CBS extrapolated level in the gas phase by AA Khairbek and M Abd Al-Hakim Badawi in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.