
Abstract
In this work, the mechanism of the palladium-catalysed acetoxylation of benzene C-H has been studied theoretically in detail. Based on experimental studies, a four-step mechanism for this reaction had been proposed, that is, C−H activation of benzene is the rate-determining step which forms an intermediate (k1 pathway) which is subsequently oxidized to produce a high-valent Pd intermediate (k2 pathway). Using quantum chemical calculations, all pathways were investigated, and the activation energy, activation enthalpy and activation Gibbs free energy for all steps were calculated and compared with each other. It was determined that the RDS proceeds through a square complex instead of a T-shaped complex. The activation energy related to the k2 pathway is higher than that of the RDS, and therefore, a new mechanism is proposed.
Introduction
At present, it is well known that palladium has greatly improved the synthesis of organic compounds, which is why Heck, Negishi and Suzuki received the 2010 Nobel Prize in Chemistry.1–3 Many reactions that seemed to be impractical in the past were made possible using palladium. 4 Therefore, understanding the mechanism of palladium-catalysed reactions is of paramount importance.5,6
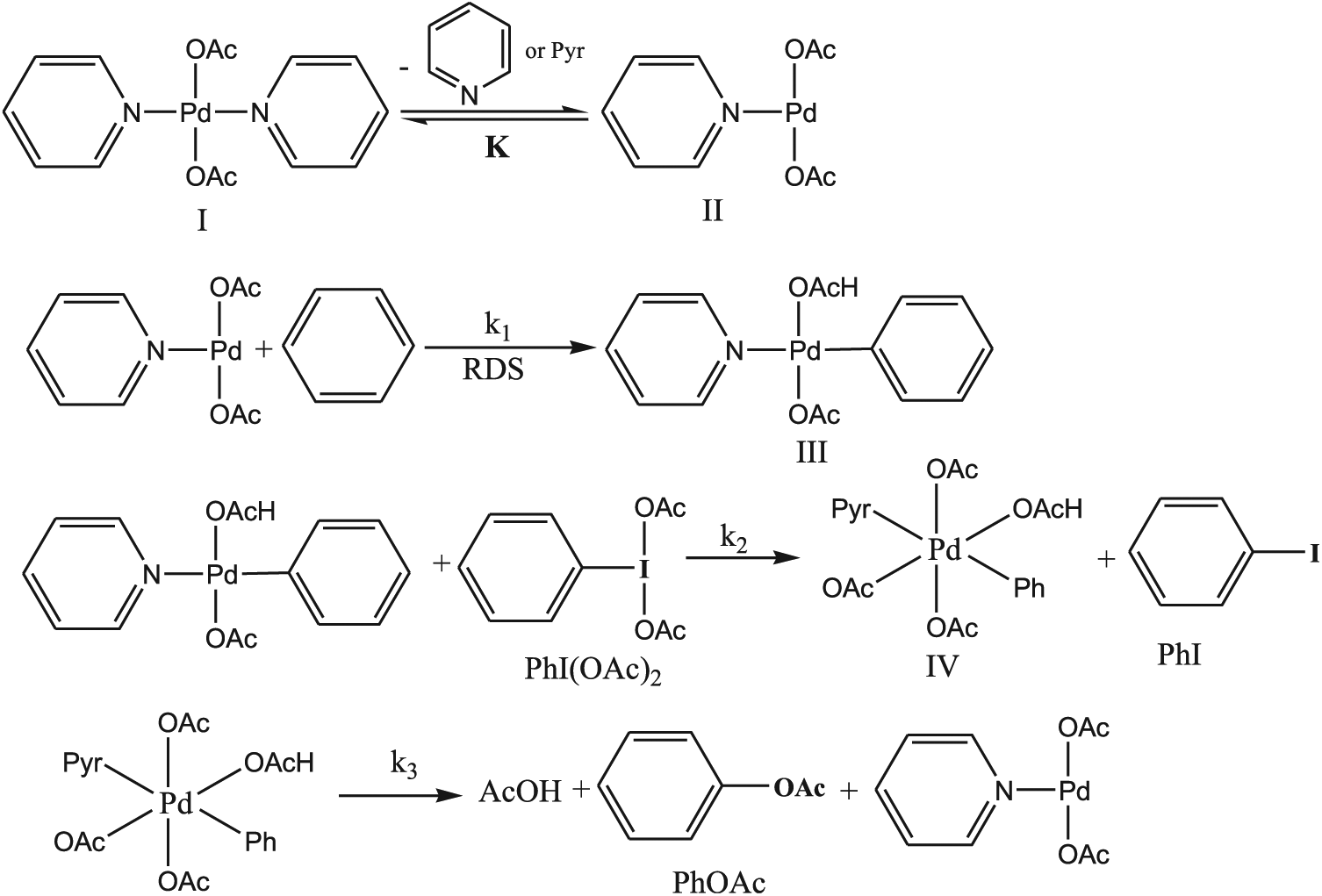
In recent years, Pd-catalysed C−H functionalization has been widely used to synthesize valuable organic compounds.7–10 There are efficient methods for direct functionalization of C−H bonds, but many of these require a directing group (DG) to increase the reactivity and selectivity of the reaction. 11 These reactions are well known mechanistically.12,13 On the other hand, non-directed Pd-catalysed C−H functionalization reactions have been less developed and mechanistically not fully understood (Scheme 1).14–16
Non-directed C–H functionalization.
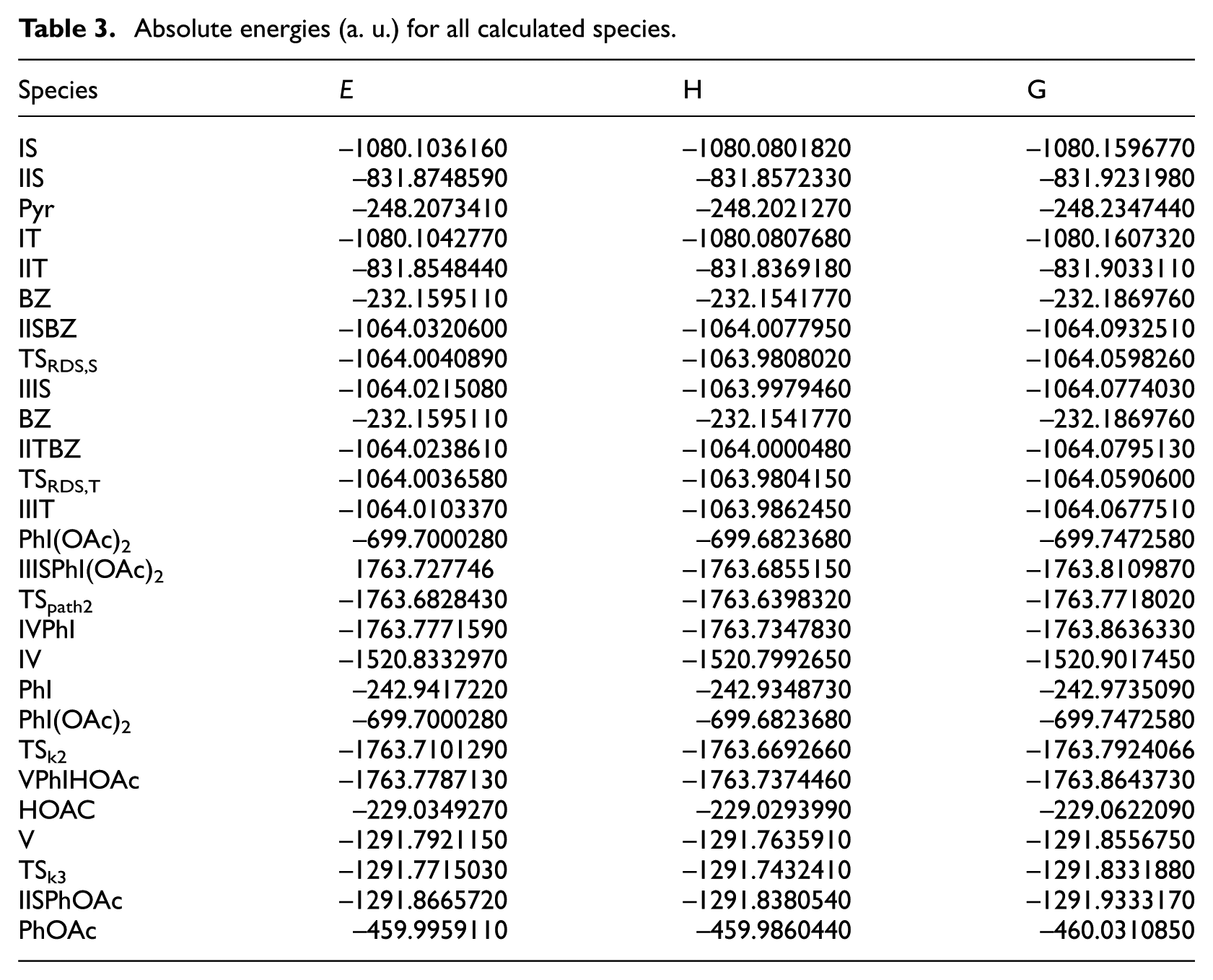
In recent years, Cook and colleagues17,18 and Emmert and colleagues19,20 have studied extensively the non-directed C−H acetoxylation of arenes. In particular, they investigated the role of pyridine (Pyr) as a supporting ligand in increasing the reaction efficiency using catalyst systems such as Pd(OAc)2(Pyr)2 and proposed the following mechanism (Scheme 2). 17
Mechanism of palladium-catalysed C−H acetoxylation of benzene.
The mechanism commences with precatalyst I (Pd(OAc)2(Pyr)2), which undergoes reversible pyridine dissociation to produce monopyridine species II. The rate-determining step (RDS) is related to C–H activation of benzene at complex II which forms intermediate III. Oxidation of this intermediate by PhI(OAc)2 forms a high-valent Pd intermediate IV. The combination of PhI(OAc)2 as oxidant and Pd(OAc)2 as catalyst was reported in 1996. 21 Intermediate IV undergoes reductive elimination to produce PhOAc and regenerate species II.
The development of appropriate molecular models is of special importance in understanding the mechanistic behaviour of catalyst systems such as Pd(OAc)2(Pyr)2. The most important method for designing such models is quantum chemical calculations. 22 In this work, we investigated the mechanism of the palladium-catalysed C−H acetoxylation of benzene using the quantum mechanical approach.
Computational method
All quantum chemical calculations have been done using the GAUSSIAN 09 package 23 at B3LYP24–26 hybrid density functional level and 6-31G(d, p) basis set. 27 For Pd and I atoms, the LANL2DZ basis set, 28 with effective core potential (ECP) functions, was used. Standard convergence criteria for geometry optimization were used. For all configurations, all degrees of freedom were optimized. The transition states obtained were confirmed to have only one imaginary frequency of the Hessian. Zero-point corrections were used to calculate activation energies. The solvent can have a significant effect on chemical systems and reactions explicitly or implicitly. The polarized continuum model (PCM) method was used to consider the implicit role of the solvent.29,30 All calculations were done in acetic acid (HOAc) as an implicit solvent.
Results and discussion
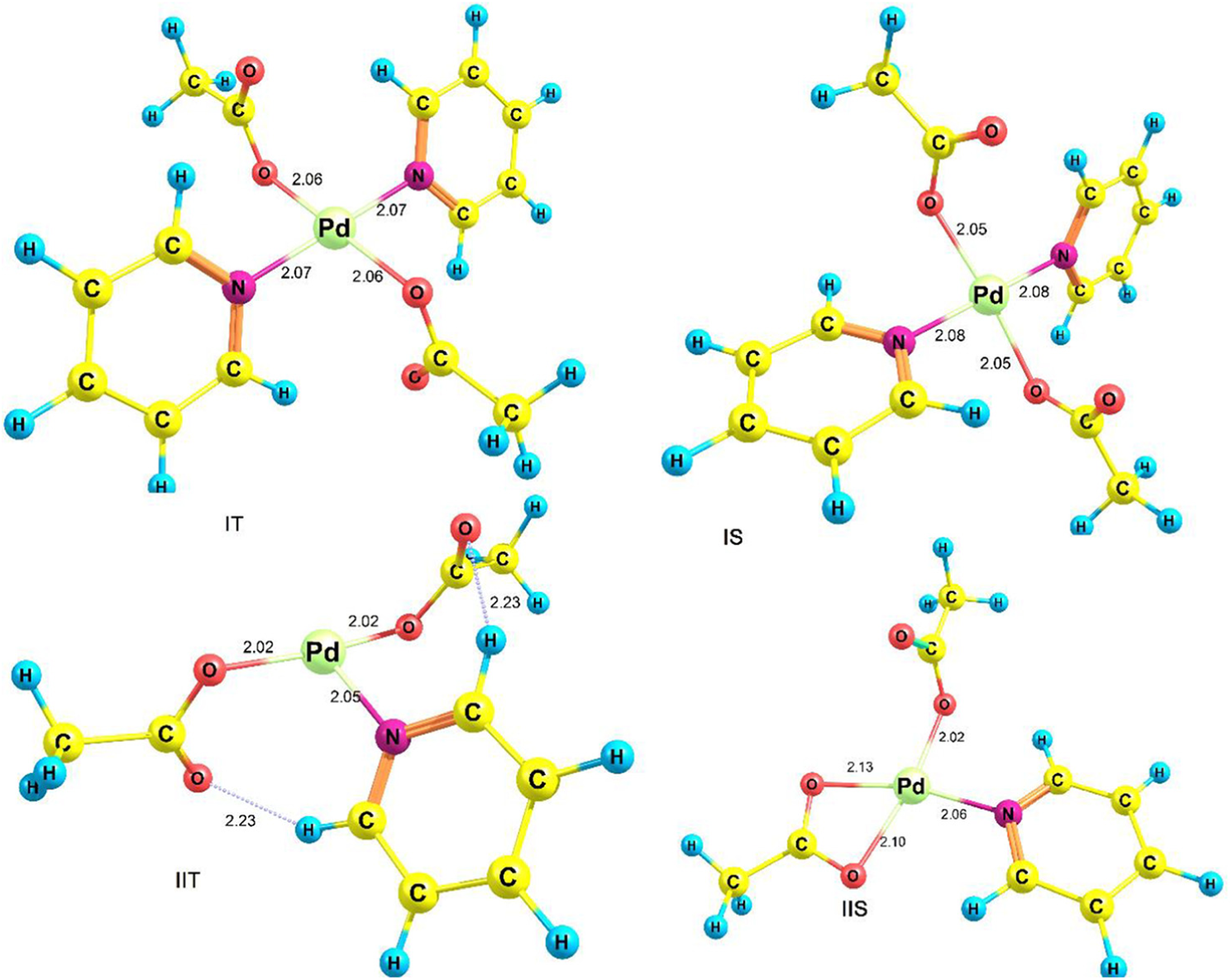
We started studying this reaction from the equilibrium between species I and II. In the study of species II, we concluded that this species has two different configurations. One configuration has a T-shaped structure that we call it IIT, and another configuration has a square structure called IIS (Figure 1). OAc is a bidentate ligand and in configuration IIS, one of the OAc ligands is coordinated to palladium via two oxygen atoms. Because the trans effect of the Pyr ring is greater than that of the OAc ligand, the CO bond length trans to the Pyr (2.13 Å) is longer than that of the OAc (2.10 Å), and the substitution of CO with benzene in the next step will occur trans to the Pyr ring (Figure 1).
Optimized structures of IT, IS, IIT and IIS.
Relative energies for the different species are presented in Table 1. In this table,
Relative energies (kJ mol–1) for all species in equilibrium (K) and RDS (k1).
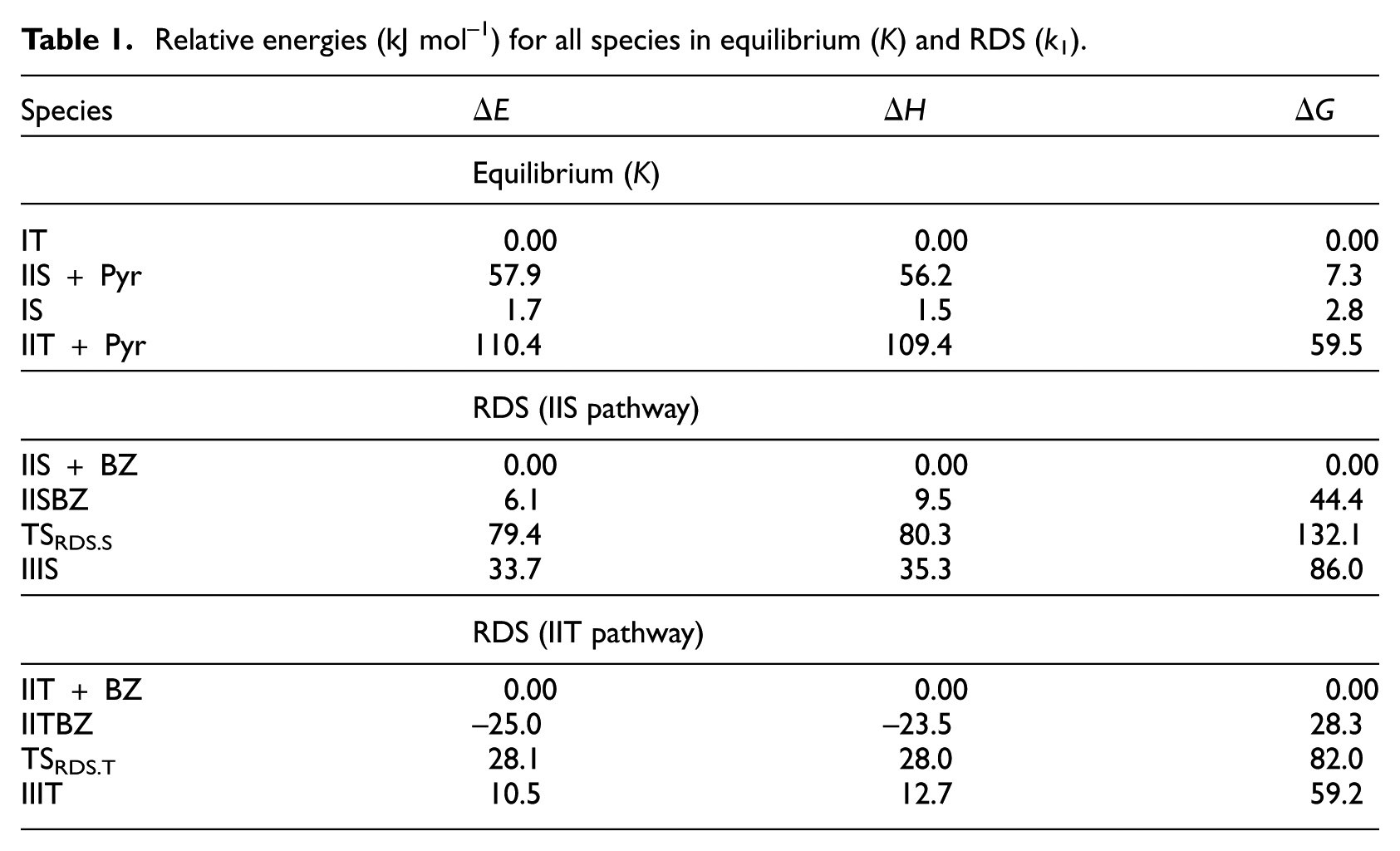
According to the two configurations IIT and IIS, there are two structures for species I referred to as IT and IS, respectively (from now on, letters T and S represent different species in the pathways related to IIT and IIS, respectively). Contrary to the configurations IIT and IIS, these two structures (IT and IS) do not differ much in energy (Table 1). Figure 1 shows the optimized structures of species IT, IS, IIT and IIS. The values of the equilibrium constant
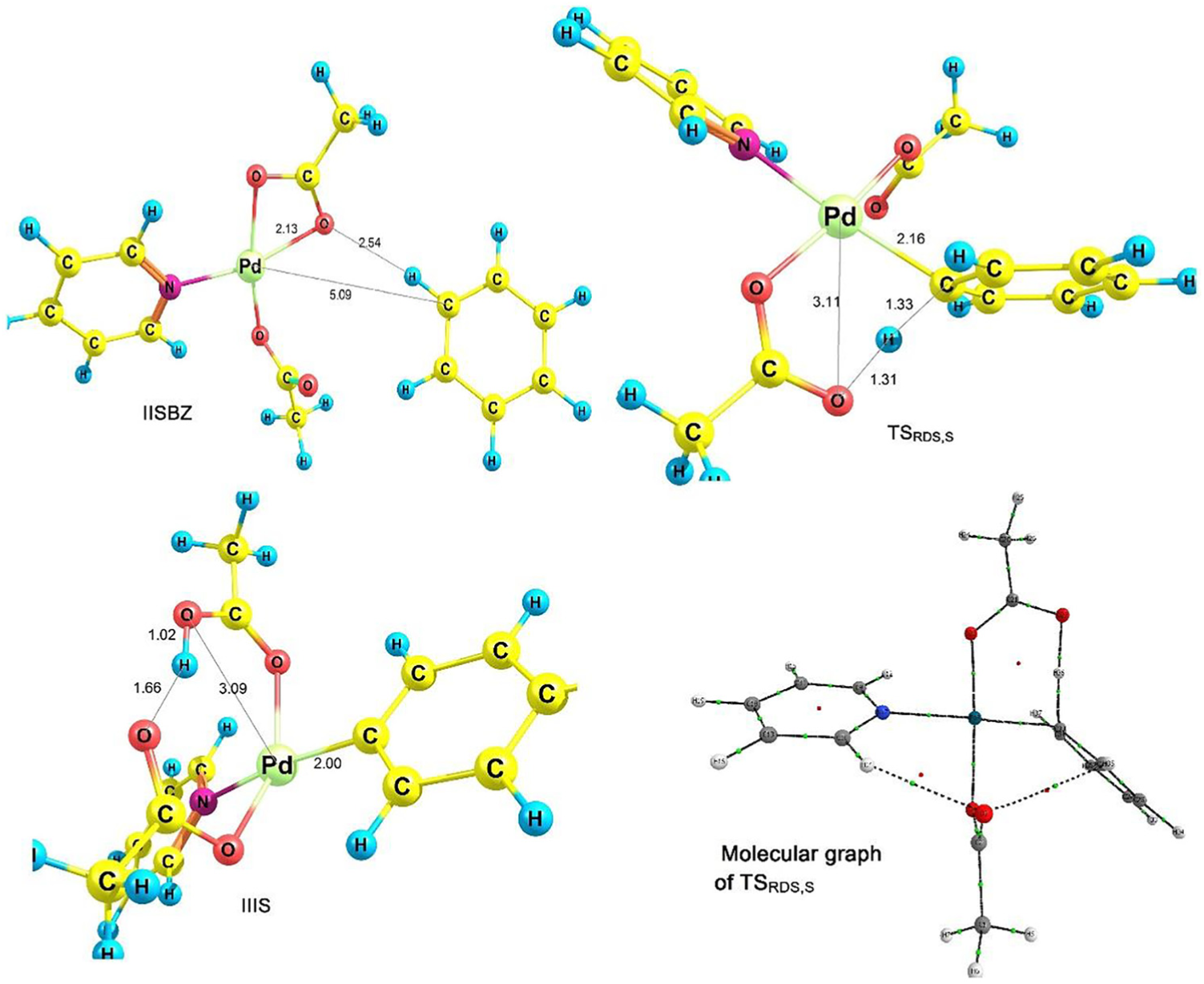
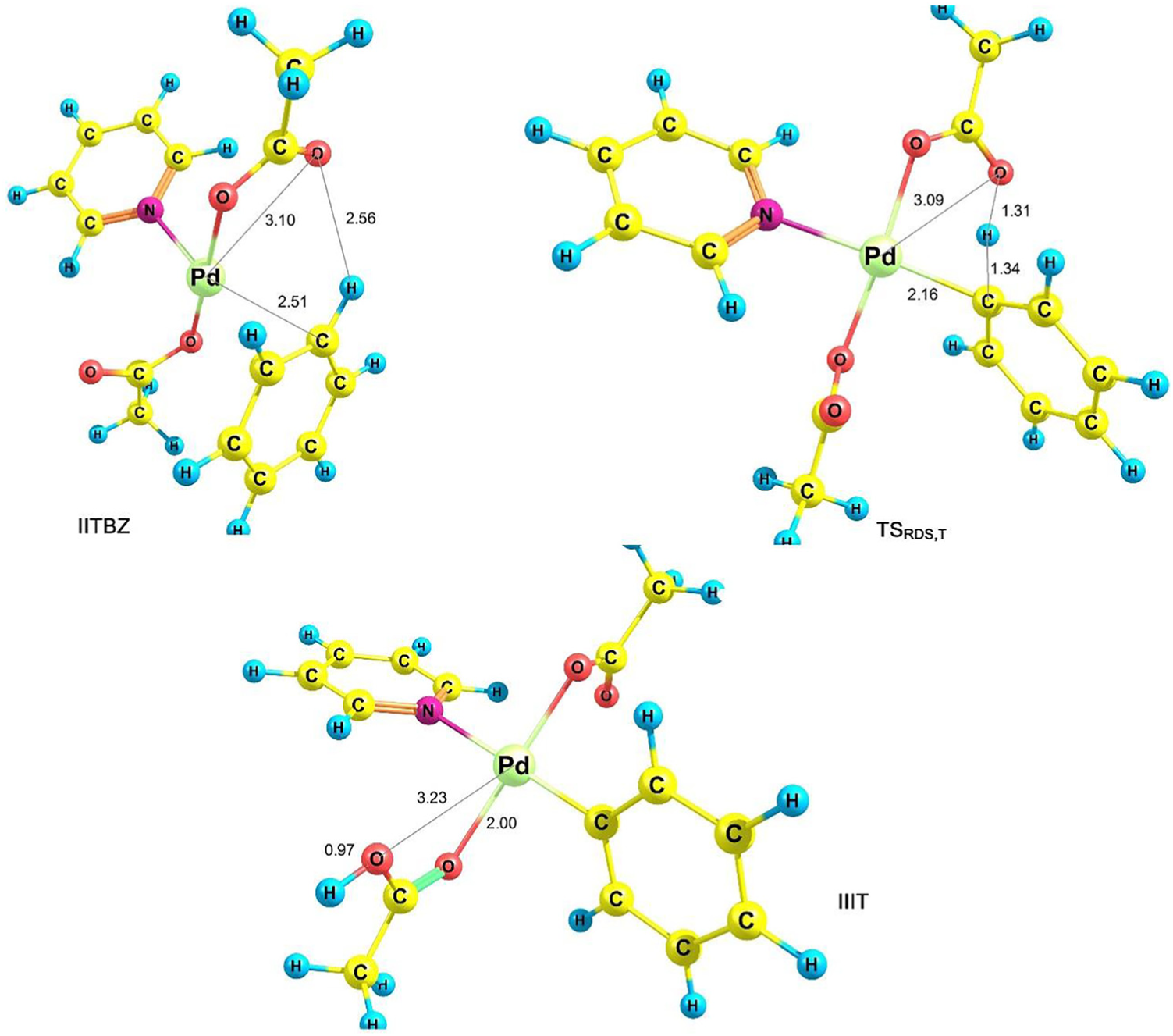
C–H activation of species II to generate species III is the RDS. In order to optimize the transition states of RDS (k1 pathway), the IIT and IIS molecules in the vicinity of benzene should be optimized (IITBZ and IISBZ in equation (1)). For species III, corresponding to IIT and IIS, two structures IIIT and IIIS are obtained. The optimized structures of IITBZ, IISBZ, IIIT and IIIS are presented in Figures 2 and 3.

Optimized structures of IISBZT, TSRDS,S and IIIS and molecular graph of TSRDS,S.
Optimized structures of IITBZT, TSRDS,T and IIIT.
Considering reactant IISBZ (IITBZ) and product IIIS (IIIT), the transition state of the RDS is obtained which we call TSRDS,S (TSRDS,T). Figures 2 and 3 present the optimized structures of TSRDS,S and TSRDS,T, respectively. Using Figure 2, the Pd-C and O-H bond lengths decrease (increase) from 5.09 and 2.54 Å (2.00 and 1.02 Å) for IISBZ (IIIS) to 2.16 and 1.31 Å for TSRDS,S, respectively.
In TSRDS,S, a six-membered transition state has been formed. Due to the fact that during this step, a proton transfer to oxygen occurs, the role of hydrogen bonds in forming the six-membered transition state is very important. For further investigation, quantum theory of atoms in molecules (QTAIMs)
31
computations were performed on TSRDS,S using the AIMALL package.
32
The values of the electron density (ρ(r)), its Laplacian
Relative energies for the different structures in the RDS are shown in Table 1. The activation energy
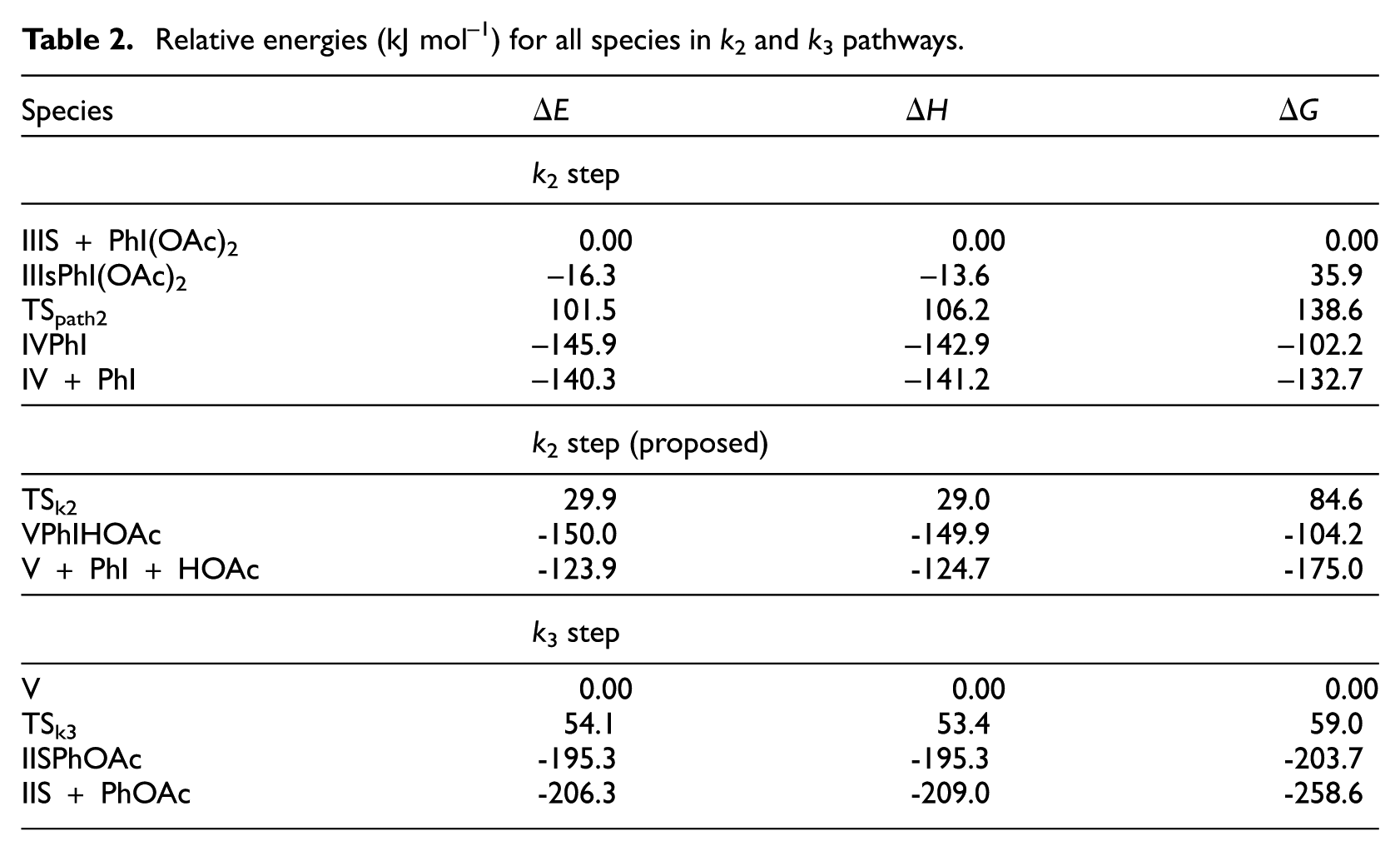
Relative energies (kJ mol–1) for all species in k2 and k3 pathways.
In step next step, a 2e oxidation of intermediate III forms species IV (equation (2))
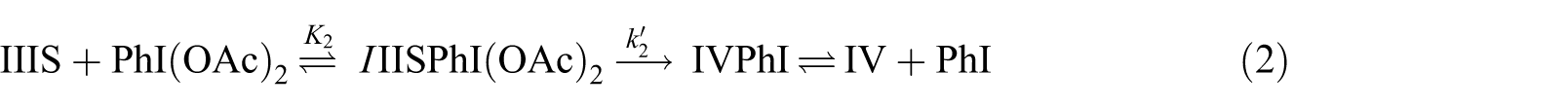
First, species IIIS and IV have been optimized in the vicinity of PhI(OAc)2 and PhI, respectively (IIISPhI(OAc)2 and IVPhI in Figure 4). Considering reactant IIISPhI(OAc)2 and product IVPhI, the transition state of
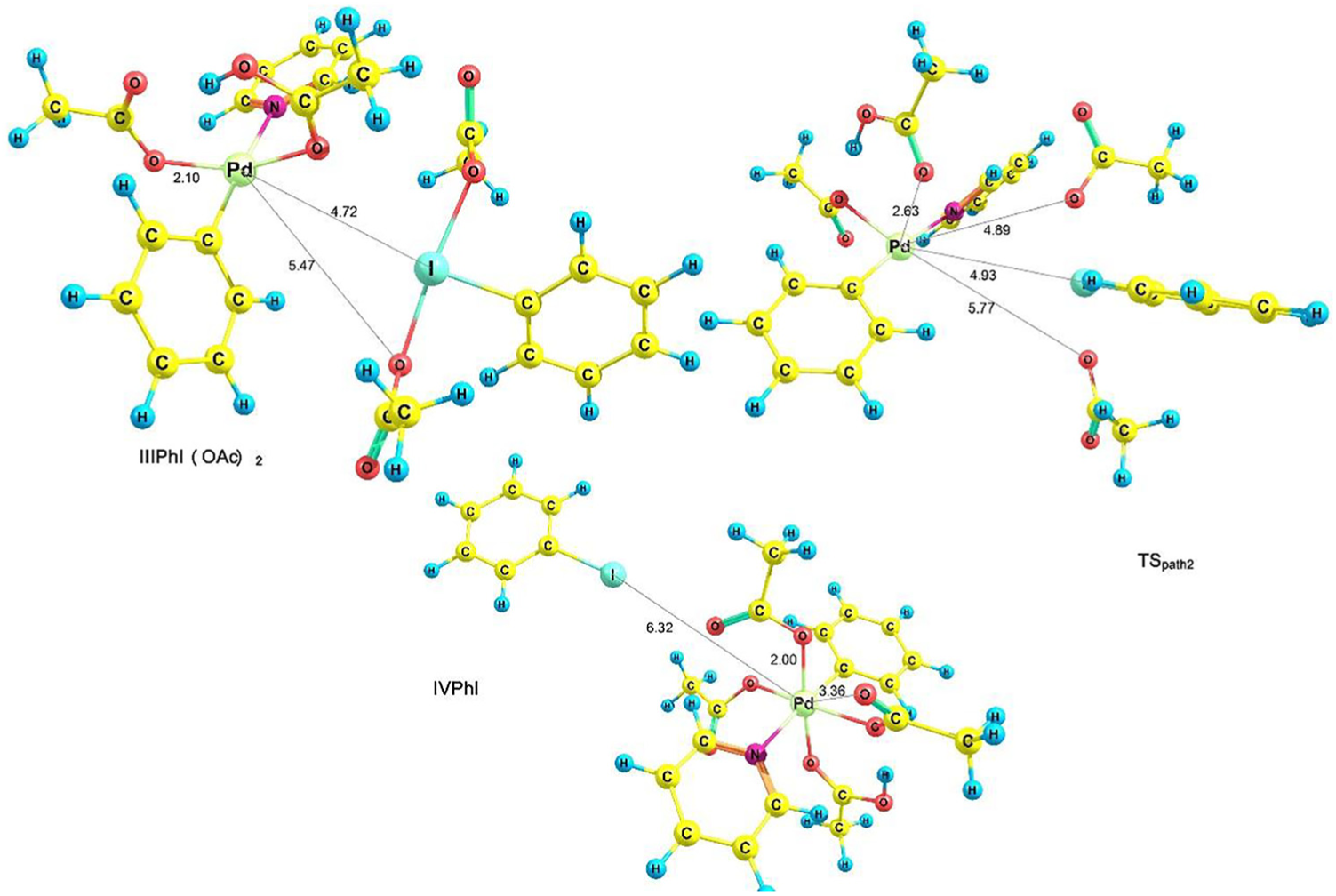
Optimized structures of IIIPhI(OAc)2, TSpath2 and IVPhI.
Figure 4 shows the optimized geometries of TSpath2, IIISPhI(OAc)2 and IVPhI. In this figure, the Pd-I bond length increases (decreases) from 4.72 (6.32) Å for IIISPhI(OAc)2 (IVPhI) to 4.93 Å for TSpath2 and the Pd-O bond length increases (decreases) from 2.10 (3.36) Å for IIISPhI(OAc)2 (IVPhI) to 2.63 Å for TSpath2. According to Table 2,
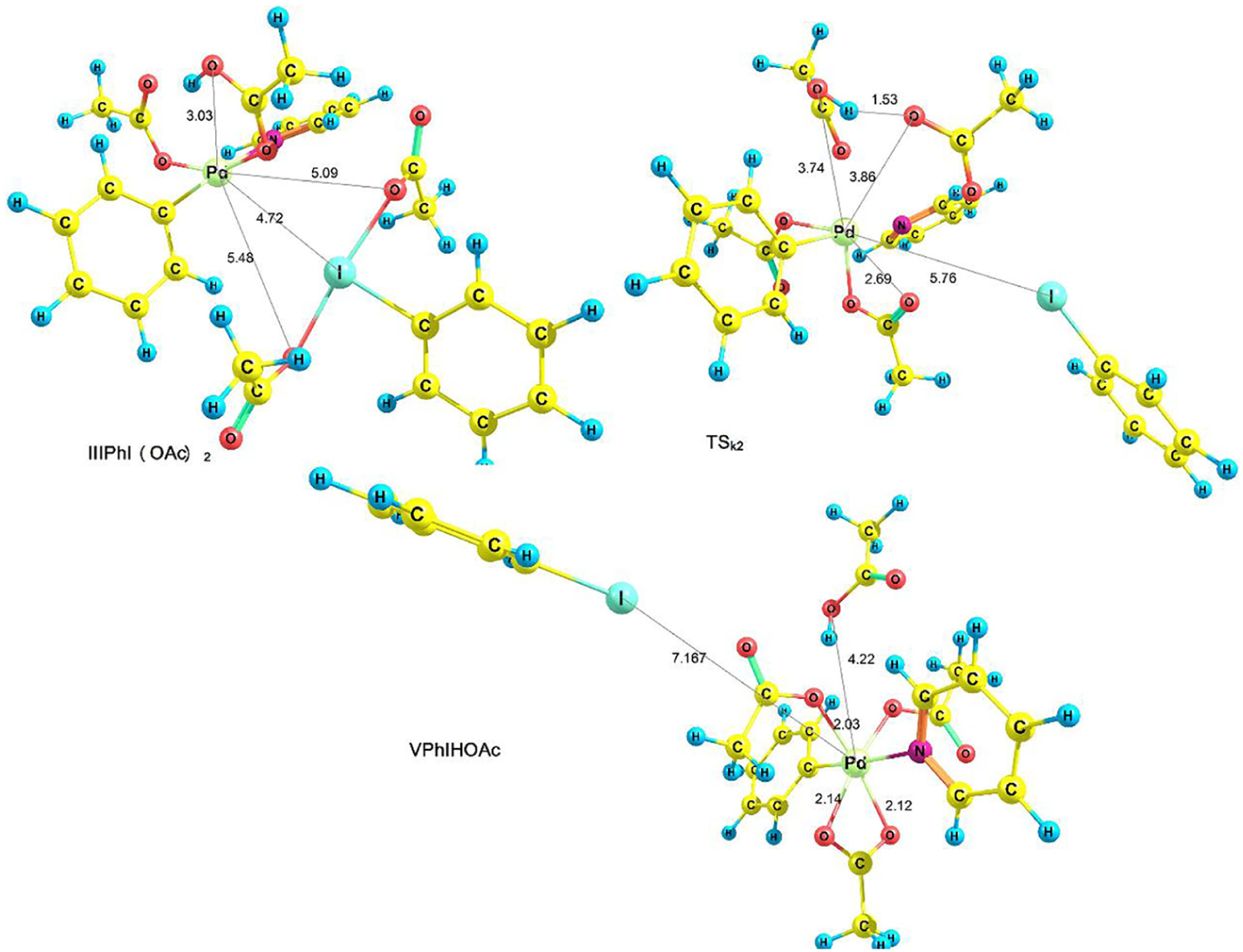
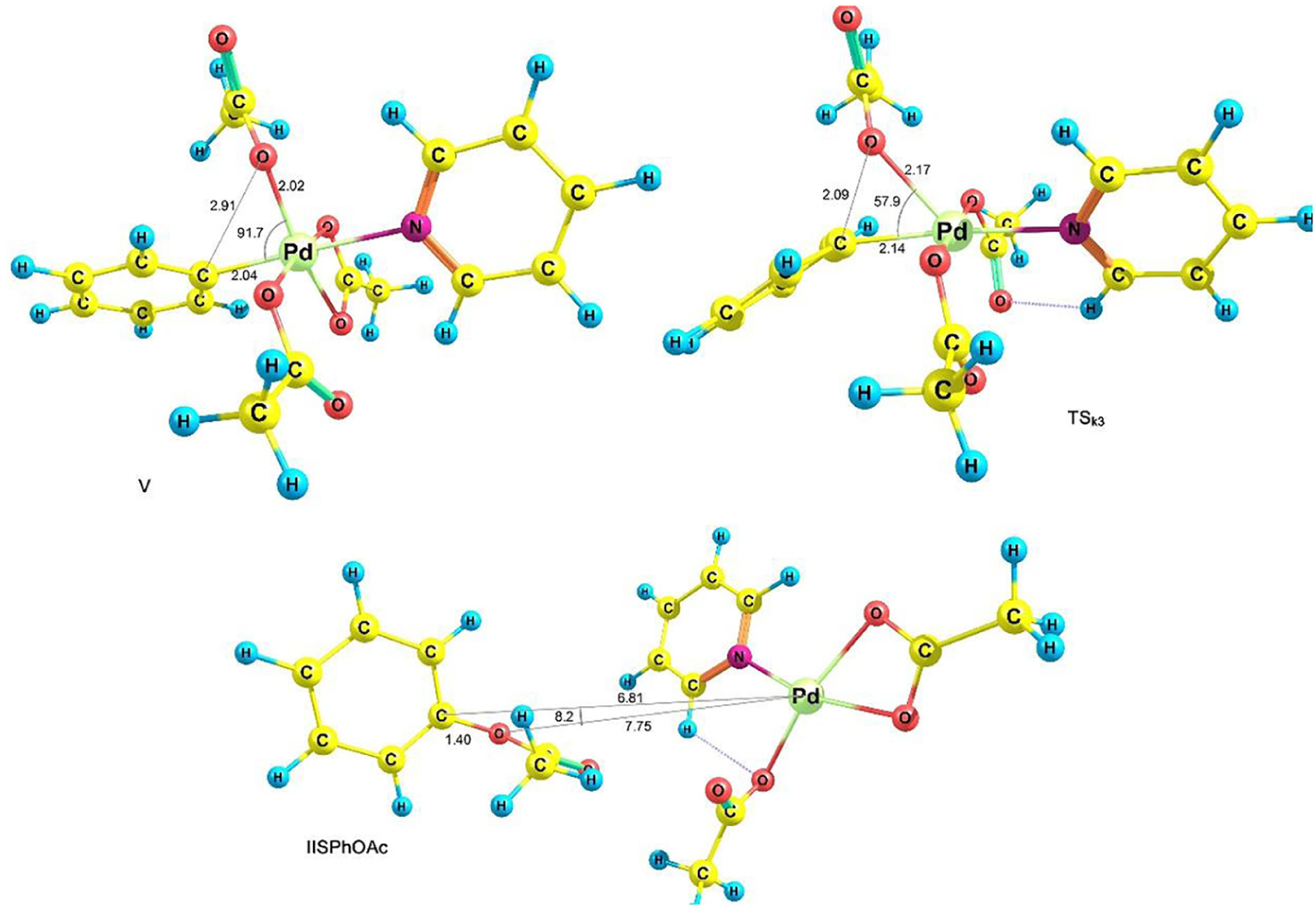
Our studies have shown that in this step, instead of the octahedral complex IV, another octahedral complex is formed (species V in Figures 5 and 6) in which one of the OAc ligands is coordinated to palladium through its two oxygen atoms, and HOAc is then released as a reaction product (equation (3))

Optimized structures of IIIPhI(OAc)2, TSk2 and VPhIHOAc.
Optimized structures of V, TSk3 and VPhIHOAc.
The transition state of step
Absolute energies (a. u.) for all calculated species.
Based on the proposed mechanism, C–O bond formation involves reductive elimination from species V which releases PhOAc and regenerates IIS at the final stage

The transition state of step
Conclusion
The mechanism of the palladium-catalysed C−H acetoxylation of benzene was investigated at B3LYP density functional level in the solution phase. This mechanism consists of four steps: (1) reversible pyridine dissociation of catalyst system (Pd(OAc)2(Pyr)2 or (I) into monopyridine species II; (2) C–H activation at the RDS (k1 pathway) and formation of intermediate III; (3) oxidation of III and formation of high-valent Pd intermediate IV (k2 pathway); (4) reductive elimination of IV (k3 pathway).
The activation energy, activation enthalpy and activation Gibbs free energy for all pathways were calculated. It was found that the RDS proceeds through a square complex instead of a T-shaped complex. The activation parameters related to k1 pathway (RDS) are lower than those of the k2 pathway. A new mechanism is proposed for the k2 and k3 pathways, and it was shown that these pathways have lower activation energies than the RDS.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.