
Abstract
Deoxygenative chlorination of carbonyl compounds using the HMe2SiCl/FeCl3/EtOAc and HMeSiCl2/FeCl3/EtOAc systems has been systemically investigated. The HMe2SiCl-FeCl3 system showed the advantages of good substrate applicability, mild reaction conditions, simple operation, low cost, and easy availability of raw materials. Also, it provided a simple and efficient synthesis route for carbonyl deoxychlorination via a one-pot method. Using the HMeSiCl2/FeCl3/EtOAc system, the β-methylchalcone derivative could be obtained in good yields in addition to obtaining the chlorinated compound. Finally, two plausible reaction routes were proposed to describe the formation of the chlorinated compound and the β-methylchalcone derivative.
Keywords

Introduction
Reduction of carbonyl compounds to hydroxyl compounds via catalytic hydrosilylation is an essential synthetic method that has been widely used in academia and industry.1–4 Since Ojima et al. 5 first discovered this reaction in 1972, an enormous amount of work has been published. The published works have mainly focused on searching for meaningful catalysts or activators that are cheap, easy-to-handle, tolerant, and chemo- and/or regioselective.6–12 However, several research groups have focused on the direct conversion of hydroxyl groups to chlorides during hydrosilylation reactions.13–18 For example, Onishi et al. 19 reported that the effective In(OH)3-catalyzed deoxygenative halogenation of carbonyl compounds using chlorodimethylsilane (HMe2SiCl) bearing both hydrogen and chlorine moieties instead of triethylsilane (HSiEt3) and chlorotrimethylsilane (Me3SiCl) as separate hydrogen and chlorine sources, respectively. Obviously, such direct conversion of carbonyl compounds into chlorides under mild conditions shows significant superiority to the conventional multistep approach involving separate reduction and chlorination under harsh conditions. Subsequently, Li et al. 20 realized a similar reaction using dichloromethylsilane (HMeSiCl2) with FeCl3 as the catalyst. In addition, the deoxygenative chlorination of aromatic carbonyl compounds has also been explored. Savela et al. 21 reported that using Fe(III) oxo acetate as a catalyst, hydrosilylation of benzylic carbonyl compounds followed by subsequent chlorination could be easily achieved with HSiEt3 (as a hydrogen source) and Me3SiCl (as a chlorine source). Savela and Leino 22 also found that if less Me3SiCl was used (e.g. 8 mol%), symmetrical and nonsymmetrical ethers were obtained.
Recently, we reported the K2CO3-activated hydrosilylation of aldehydes and ketones with inexpensive polymethylhydrosiloxane as a reductive reagent. 23 In this work, we expanded our strategy to the deoxygenative halogenation of carbonyls using silicon reagents and inexpensive catalysts. Herein, we present FeCl3-catalyzed deoxygenative chlorination of carbonyl compounds using HMe2SiCl or HMeSiCl2 as the hydride and chloride sources, respectively, in an efficient and simple manner.
Results and discussion
HMe2SiCl as hydride and chloride sources
First, acetophenone was used as a standard substrate, and the catalytic activity of various catalysts was studied (Table 1). Ethyl acetate (EtOAc) was used as the solvent, and the deoxygenative chlorination could not be conducted without catalyst (Table 1, entry 1). However, high conversion was obtained when the reaction was carried out in the presence of Lewis acids (Table 1, entries 2–8). Common Lewis acids, such as Fe(C2H3O2)2, Fe(ClO4)3·3H2O, FeCl2, In(OH)3, Fe(acac)3, AlCl3, and FeCl3, all exhibited excellent catalytic activity. When FeCl3 was used, 97% yield of chloroethylbenzene was obtained within 2 h (Table 1, entry 8). Because FeCl3 is one of the least expensive inorganic catalysts, other catalysts were not investigated in subsequent studies. To further optimize the reaction, the molar ratios of HMe2SiCl to acetophenone varied (Table 1, entries 9–11). The results indicate that yields of the corresponding chlorides decreased with a decrease in the amount of HMe2SiCl (Table 1, entries 10 and 11). When diminishing the equivalent of HMe2SiCl to 1.0, the yield of the chloroethylbenzene was only 73% after 24 h (Table 1, entry 11). Notably, the 1.8 equiv. of HMe2SiCl could not lead to the yield improvement, while the reaction time could be reduced to 1 h (Table 1, entry 9).
Optimized reaction conditions. a


DME: 1,2-dimethoxyethane; DMF: N,N-dimethylformamide; DCM: dichloromethane.
All reactions were carried out with
Yields were determined by 1H NMR.
The bold values in Table 1 are used to highlight that this is the optimal reaction condition.
The influences of solvents on deoxygenative chlorination were also investigated (Table 1, entries 12–19). When 1,4-dioxane was used as the solvent, the desired compounds were only obtained in 66% yield (Table 1, entry 12), while 1,2-dimethoxyethane (DME) led to an even lower yield of 44% (Table 1, entry 13). For other solvents, such as acetonitrile (MeCN), ether (Et2O), hexane, and N,N-dimethylformamide (DMF), the starting substrate was fully recovered or only a trace amount of the desired compound was obtained (Table 1, entries 14–17); this indicates that no deoxygenative chlorination occurred. When dichloromethane (DCM) and chloroform were used, some side products were generated, and the desired compound was not detected (Table 1, entries 18 and 19). Although it was reported that the carbonyl substrates gave good yield of the chloro compounds in chloroform using HMe2SiCl with In(OH)3 as catalyst, 19 using chloroform as solvent in our experiment, we could only recover most of the starting material. Thus, EtOAc is the preferred solvent for the deoxygenative chlorination reaction, and this is probably because the good solubility of FeCl3 in EtOAc and the involving of a solvent-coordinated hexavalent silicate which acts as an active hydride species.
After the reaction conditions were optimized, various ketones were tested as substrates for the deoxygenative chlorination reaction (Table 2). Unsubstituted aromatic ketones gave corresponding chlorides in high yields (Table 2, entries 1–3). While the aromatic ketones that had a deactivating substituent (Table 2, entries 4–17) and those that had a weak-activating substituent (Table 2, entries 18–20) were converted to chlorides in good-to-excellent yields (79%–100%); the substituents susceptible to reduction such as MeSO2– (Table 2, entry 10), NO2– (Table 2, entries 11 and 12), NC– (Table 2, entry 13), CH3OOC– (Table 2, entry 14), and HOOC– (Table 2, entry 15) were not affected. For the acetophenones that had the same substituent at different positions, the yields were similar (Table 2, entries 4 and 5; entries 6 and 7; entries 11 and 12). However, each aromatic ketone that had an activating substituent (Table 2, entries 21–23) failed to give the desired chloride, and the Clemmensen-type reduction product was obtained. These results agree well with Onishi’s report, in which chloroform was used as the solvent. 19 For aromatic ketones that had an activating substituent, the reaction conditions were further optimized to obtain the desired chloride.
Substrate scope of different carbonyl compounds. a

Reaction condition:
The values below the structures refer to NMR yields.
30.0 mmol of HMe2SiCl was used.
Reaction carried out at 77 °C, 2-chloroheptane (
At room temperature, the equivalent of the HMe2SiCl was increased to 2 or 3, and no target chloride was obtained, whereas the yield of the reduction product (alkyl benzene) increased. With subsequent heating to 77 °C, the target chloride was still not obtained. Polycyclic aromatic substrates (Table 2, entries 24 and 25) also effectively produced the desired chloride. Benzophenone (Table 2, entry 26) produced chloride in 54% yield along with diphenylmethane in 43% yield. When aliphatic ketones are used as the substrates, the yields of the products were medium to high (Table 2, entries 27–29); sometimes higher temperature was necessary to obtain a satisfactory yield (Table 2, entry 30). In this context, aromatic heterocyclic ketones, such as thiophene-2-ketone (Table 2, entry 31), could not be converted to the corresponding chloride, although a trace amount of the Clemmensen-type reduction product was obtained.
HMeSiCl2 as hydride and chloride sources
Sheng et al. 24 reported that FeCl3-catalysted deoxygenative chlorination of acetophenone can also be performed using HMeSiCl2 as hydride and chloride sources with DME as the solvent. Following their report, we used p-chlorobenzophone as the substrate in our experiment. Surprisingly, a β-methylchalcone derivative was obtained as the major product in addition to the desired chloride. Also, there existed a trace amount of 4-chlorostyrene. Subsequently, EtOAc was used as the solvent to further study this reaction and the results are summarized in Table 3. Aromatic ketones that had a deactivating substituent (Table 3, entries 1 and 2) afforded β-methylchalcone derivatives along with the chlorinated compound. Aromatic ketones that had a weak-activating substituent (Table 3, entries 3 and 4) resulted in only β-methylchalcone derivatives in 82% and 72% yields, respectively, and no chloride was obtained. However, aromatic ketones that had an activating substituent, such as methoxy and hydroxy (Table 3, entries 5 and 6), failed to give β-methylchalcone derivatives and chlorides. These results agree with results reported by Elmorsy et al., 25 who treated tetrachlorosilane with aromatic ketones; the substituents that increased the negativity of the acetyl group decreased the yield of β-methylchalcone derivatives.
Reaction under the HMeSiCl2/FeCl3/EtOAc system. a


Reaction condition:
The values below the structures refer to NMR yields.
We also found that no allyl chloride could be detected, implying that β-methylchalcone derivative could not be deoxychlorinated in the HMeSiCl2/FeCl3/EtOAc system. Aliphatic ketones (Table 3, entries 7–9) failed to give the desired products at room temperature, and almost all of the starting arenes were recovered. These results imply that HMeSiCl2 probably reacted in a very different role than HMe2SiCl in the deoxygenative chlorination reaction.
Mechanism proposal
On the basis of the above results, two plausible reaction routes were proposed to explain the formation of the chlorinated compound and β-methylchalcone derivative (Scheme 1). In 2002, Onishi et al. 19 reported that deoxyhalogenation of carbonyl compounds with HMe2SiCl was catalyzed by In(OH)3 and gave the proposed mechanism. We believe that the HMe2SiCl/FeCl3 and HMeSiCl2/FeCl3 systems in our experiment each gave the chlorinated compound in a similar way to that proposed by Onishi (Scheme 1, path A). However, when HMeSiCl2 is employed as both the hydride and chloride sources, alcoholysis of HMeSiCl2 should be considered. Alcoholysis of organochlorosilane and alcohols is an important chemical characteristic of the Si–Cl bond. 26 HMeSiCl2 is more electrophilic than HMe2SiCl because of one more chlorine atom bonded to silicon. When HMeSiCl2 is used, the enol form of ketones (nucleophile) can easily attack dichloromethylsilane to give enol silyl ether (Scheme 1, path B). First, alcoholysis of the enol form of ketones occurs and affords an enol silyl ether. Then, nucleophilic attack of enol silyl ether on another ketone under the activation of FeCl3 produces an intermediate chelate via loss of dichloromethylsilane. Finally, the chelate collapses to produce the final β-methylchalcone derivative via loss of FeCl3 and H2O.
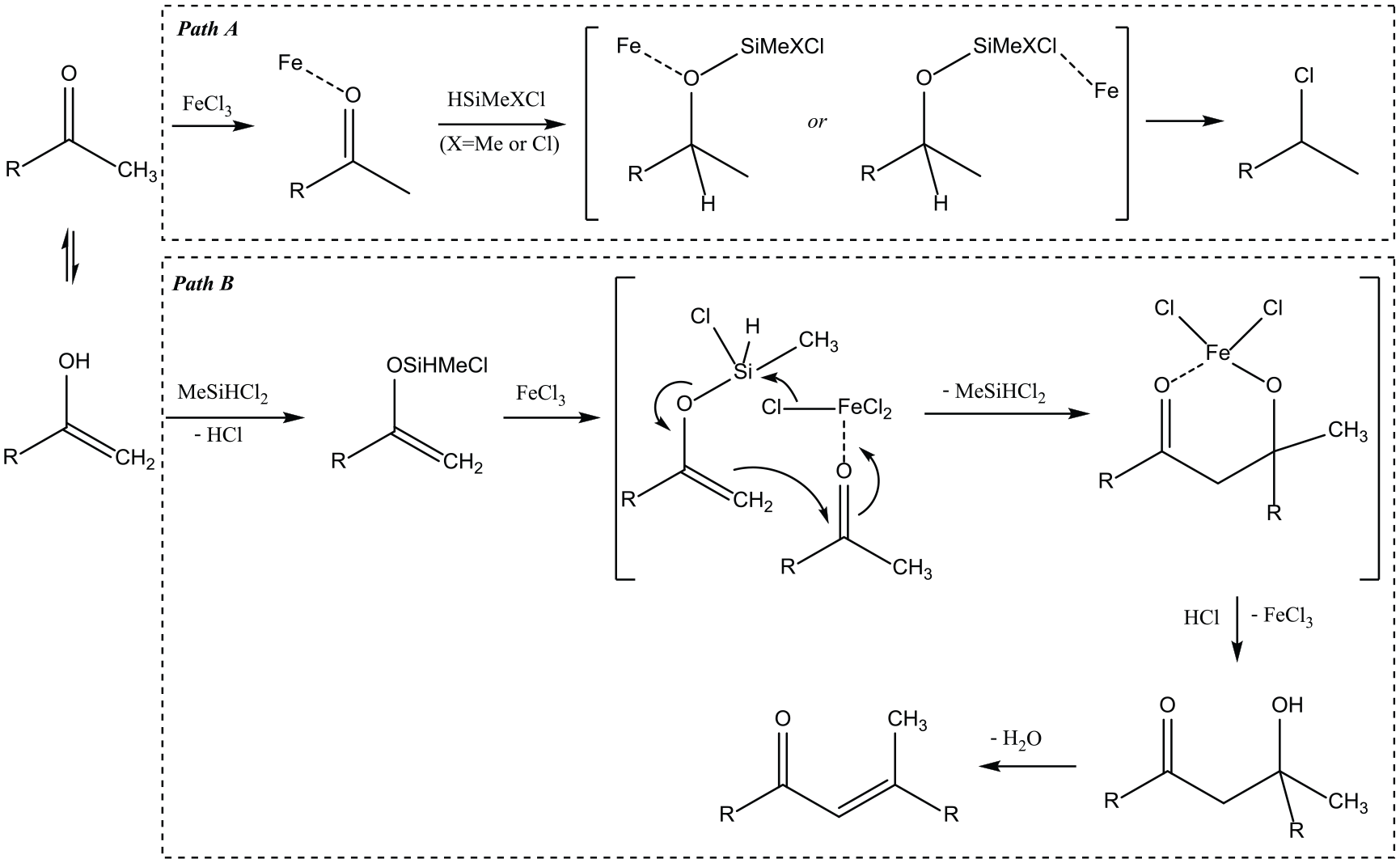
Possible routes for the formation of chlorinated compound and β-methylchalcone derivative.
Conclusion
In summary, we have achieved deoxygenative halogenation of carbonyl compounds using the HMe2SiCl/FeCl3/EtOAc system. This method is eco-friendly and shows good tolerance. We also performed deoxygenative halogenation using the HMeSiCl2/FeCl3/EtOAc system, and β-methylchalcone derivative was obtained in high yields in addition to obtaining the target chlorinated compound. The differences between the HMe2SiCl/FeCl3/EtOAc and HMeSiCl2/FeCl3/EtOAc systems were investigated and the corresponding mechanisms were proposed. We propose that the chlorinated compound was given in a similar way to the mechanism proposed by Onishi for both the systems, while a possible route, involving the alcoholysis of enol form of ketone followed by nucleophilic attack on another ketone, was proposed for the formation of the β-methylchalcone derivative in the HMeSiCl2/FeCl3/EtOAc system.
Experimental
General information
Chemicals were purchased from commercial sources and used without further purification unless otherwise stated. Deuterochloroform (CDCl3) and HMe2SiCl were purchased from J&K Scientific Ltd. All of the other chemicals were purchased from Beijing InnoChem Science & Technology Co., Ltd., Acros, and Alfa Aesar. Column chromatography was performed using 100–200 mesh silica gel, which was purchased from Qingdao Haiyang Chemical Co., Ltd. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker 400 MHz spectrometer (1H: 400 MHz; 13C: 101 MHz).
General procedure for the deoxygenative chlorination of carbonyl compounds with HMe2SiCl
A 50 mL single-neck round-bottom flask was loaded with carbonyl compound (10.0 mmol), FeCl3 (0.0811 g, 0.5 mmol), and EtOAc (20 mL). The reaction mixture was stirred at room temperature for 1 min, and then HMe2SiCl (1.4193 g, 15.0 mmol) was added. Subsequently, the reaction flask was equipped with a 90° glass joint with a balloon to protect the mixture from moisture. The reaction mixture was stirred vigorously at room temperature until the reaction was completed (as detected by thin-layer chromatography (TLC)). The solution was washed with saturated NaHCO3 solution (3 × 10 mL) to remove FeCl3. The organic layer was dried over anhydrous Na2SO4. The solvent was then removed under vacuum, and the mixture was purified via column chromatography to afford the product.
General procedure for the deoxygenative chlorination of carbonyl compounds with HMeSiCl2
A 50 mL single-neck round-bottom flask was loaded with carbonyl compound (10.0 mmol), FeCl3 (0.0811 g, 0.5 mmol), and EtOAc (20 mL). The reaction mixture was stirred at room temperature for 1 min, and then HMeSiCl2 (1.7255 g, 15.0 mmol) was added. Subsequently, the reaction flask was equipped with a 90° glass joint with a balloon to protect the mixture from moisture. The reaction mixture was stirred vigorously at room temperature until the reaction was completed (as detected by TLC). The solution was washed with saturated NaHCO3 solution (3 × 10 mL) to remove FeCl3. The organic layer was dried over anhydrous Na2SO4. The solvent was then removed under vacuum, and the mixture was purified via column chromatography to afford the product.
Characterization of the products 4a -5t
(1-Chloroethyl)benzene (
(1-Chloropropyl)benzene (
(Chloro(cyclohexyl)methyl)benzene (
1-(1-Chloroethyl)-4-fluorobenzene (
1-(1-Chloroethyl)-3-fluorobenzene (
1-Chloro-2-(1-chloroethyl)benzene (
1-Chloro-4-(1-chloroethyl)benzene (
1-Bromo-4-(1-chloroethyl)benzene (
1-(1-Chloroethyl)-4-(trifluoromethyl)benzene (
1-(1-Chloroethyl)-4-(methylsulfonyl)benzene (
1-(1-Chloroethyl)-4-nitrobenzene (
1-(1-Chloroethyl)-3-nitrobenzene (
4-(1-Chloroethyl)benzonitrile (
Methyl 4-(1-chloroethyl)benzoate (
4-(1-Chloroethyl)benzoic acid (
1,3-Bis(1-chloroethyl)benzene (
1-Chloro-3-(1-chloropropyl)benzene (
1-(1-Chloroethyl)-2-methylbenzene (
1-(1-Chloroethyl)-4-methylbenzene (
4-(1-Chloroethyl)-1,1′-biphenyl (
1-Ethyl-4-methoxybenzene (
4-Ethylphenol (
N-(4-ethylphenyl)acetamide (
2-(1-Chloroethyl)naphthalene (
9-Chloro-9H-fluorene (
(Chloromethylene)dibenzene (
(3-Chlorobutyl)benzene (
Chlorocyclohexane (
2-Chloroheptane (
Diphenylmethane (
1-(Heptan-2-yloxy)-2-methylheptane (
1,3-Bis(4-chlorophenyl)but-2-en-1-one (
1,3-Bis(4-nitrophenyl)but-2-en-1-one (
1,3-Di-p-tolylbut-2-en-1-one (
1,3-Di([1,1′-biphenyl]-4-yl)but-2-en-1-one (
Supplemental Material
Supplementary_Material – Supplemental material for Ferric chloride–catalyzed deoxygenative chlorination of carbonyl compounds: A comparison of chlorodimethylsilane and dichloromethylsilane system
Supplemental material, Supplementary_Material for Ferric chloride–catalyzed deoxygenative chlorination of carbonyl compounds: A comparison of chlorodimethylsilane and dichloromethylsilane system by Bing-Han Xing, Xuan-Xuan Zhao, Yu-Jun Qin, Pu Zhang and Zhi-Xin Guo in Journal of Chemical Research
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this paper.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this paper: This work was supported by the Fundamental Research Funds for the Central Universities and the Research Funds of Renmin University of China (Grant No. 15XNLQ04).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.