A novel synthetic route toward vilazodone is described by using 4-cyanoaniline and 5-bromo-2-hydroxybenzaldehyde as starting materials, with an overall yield of 24% and 99% purity. First, the intermediate (3-(4-chlorobutyl)-1H-indole-5-carbonitrile) is synthesized via diazotization of 4-cyanoaniline, followed by Fischer indole cyclization with 6-chlorohexanal. Subsequently, another intermediate, 5-(piperazin-1-yl)benzofuran-2-carboxamide, is generated via aromatic nucleophilic substitution of 5-bromobenzofuran-2-carboxamide with piperazine. Finally, vilazodone is obtained via nucleophilic substitution of the above two key intermediates by treatment with Et3N/K2CO3. In comparison to the original process, this route avoids the use of expensive and toxic reagents and resolves issues such as safety, environmental concerns, and high costs.
Vilazodone (7), a new type of antidepressant drug, is widely used for the treatment of major depression in adults. To date, several efficient methods for synthesis of vilazodone have been reported. For example, in 2004, Timo Heinrich’s group described a synthetic route for the preparation of vilazodone by using 5-cyanoindole and 5-nitrobenzofuran-2-carboxamide as starting materials, with an overall yield of 10%–20% being obtained.1,2 First, 5-cyanoindole (1) was reacted with 4-chlorobutanoyl chloride under catalysis by isobutyl-AlCl2 to afford the chloride 2, which was then reduced to 3-(4-chlorobutyl)-1H-indole-5-carbonitrile (3) by treatment with 2-(methoxyethoxy)aluminum hydride. Subsequently, 5-nitrobenzofuran-2-carboxamide (4) was reduced to compound 5 via hydrogenation with Pd/C and, then, further transformed into 5-(piperazin-1-yl)benzofuran-2-carboxamide (6) by reaction with bis(2-chloroethyl)amine. Vilazodone (7) was finally obtained by through nucleophilic substitution of compounds 3 and 6 with K2CO3/DMF (Scheme 1). This synthetic route employed many expensive and toxic reagents, leading to environmental and high cost issues. Subsequently, Xu’s group provided another synthetic route toward compound 3 (Scheme 2). 5-Cyanoindole (1) was protected using TsCl to give compound 8, which was then reacted with 4-chlorobutanoyl chloride under catalysis of AlCl3 to afford compound 9 in 90% yield. Compound 9 was reduced to compound 10 by treatment with NaBH4/CF3COOH in 95% yield.3 Finally, 3-(4-chlorobutyl)-1H-indole-5-carbonitrile (3) was obtained via hydrolysis of compound 10. Vilazodone (7) was obtained via nucleophilic substitution reaction of compounds 3 and 6, using the same method as Tim Heinrich’s group.1–3
Current synthesis of vilazodone (7).
Current optimal synthesis of 3-(4-chlorobutyl)-1H-indole-5-carbonitrile (3).
The two above-mentioned synthetic routes both involve environmentally unfriendly reagents,4,5 such as bis(2-chloroethyl)amine and NaBH4. In addition, 5-cyanoindole (1) and 5-nitrobenzofuran-2-carboxamide (4) were also demonstrated to be expensive and difficult to synthesize. Herein, we describe a novel synthetic route toward vilazodone (7) that is more suitable for industrial production. This route is advantageous due to its low cost and high yield.
Results and discussion
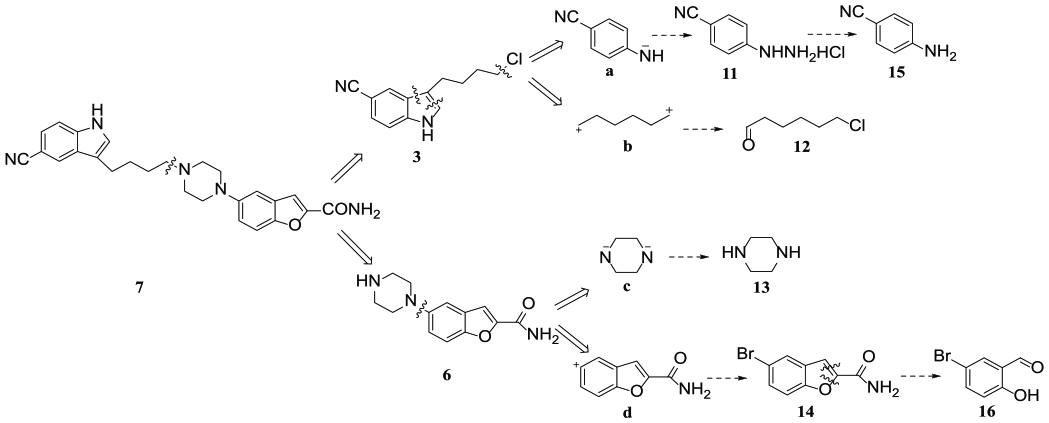
A retrosynthesis of the new synthetic route is shown in Scheme 3. Vilazodone (7) can be disconnected into intermediates 3 and 6. These intermediates 3 and 6 would give four fragments a, b, c, and d, and these fragments can be disconnected into compounds 11, 12, 13, and 14. Compound 11 can be synthesized through diazotization and reduction of amine 15, and compound 14 can be synthesized via cyclization of aromatic aldehyde 16 with BrCH2COOCH2CH3.
The retrosynthesis of vilazodone (7).
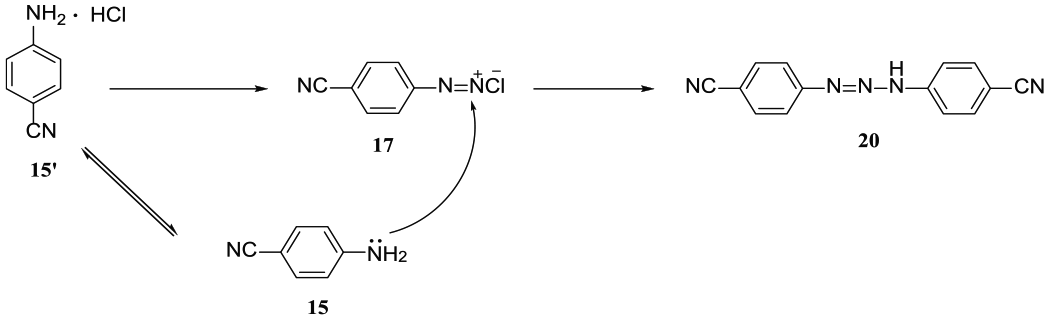
There have been several routes reported for synthesis of compound 3.6–8 One of the most suitable routes, which was developed by Xu’s group,3 employed 1H-indole-5-carbonitrile (1) as the starting material. We have developed an efficient synthetic route to prepare compound 3 (Scheme 4), which is inexpensive and uses commercially available reagents. First, because of the strong electron-withdrawing substituent, it proved difficult to convert 4-aminobenzonitrile (15) into the corresponding salt with HCl. In order to increase the yield of 4-cyanobenzenediazonium hydrochloride (17), 3 equiv. of HCl and 1.1 equiv. of NaNO2 were required, and the extent of conversion of 4-aminobenzonitrile (15) was almost 90%. However, the by-product 20 (Scheme 5) was also observed during this process.9 To decrease the formation of 20, the effect of the reaction temperature on this reaction was investigated. Fortunately, it was observed that increasing the reaction temperature reduced the quantity of by-product 20, and a temperature of 2°C proved to be the best choice (Figure 1). Subsequently, 4-cyanophenylhydrazine hydrochloride (11) was easily obtained in 90% yield via reduction of 4-cyanoaniline diazonium salt 17 with NaHSO3 at 85°C for 2 h (Scheme 4). However, there existed a problem in which a large quantity of HCl (36.5%, triple the volume of the reaction solution) was needed to salt out compound 11, which resulted in a lot of waste acid. In order to solve this problem, the HCl was recycled at least four times. The results indicated that the yield of compound 11 using recycled HCl four times was almost 80% and was reduced markedly after more than five times (Figure 2).
The efficient synthesis of the key intermediate 3.
The formation of by-product 20.
Change in the yield of by-product 20 based on the reaction temperature during diazotization. Reaction conditions: 1 equiv. of 4-aminobenzonitrile, 3 equiv. of HCl, 1.1 equiv. of NaNO2, 1 h.
Change in the yield of compound 11 with recycled times of HCl during salting out. Recycled HCl was used directly without any purification.
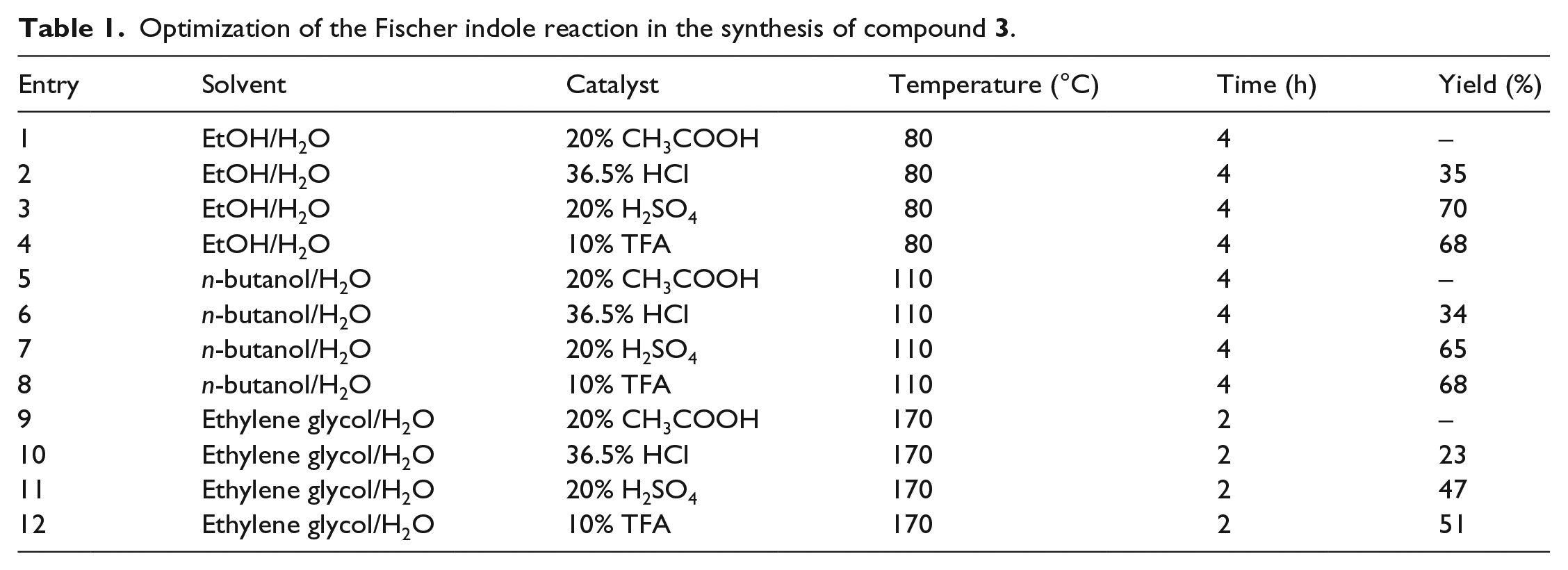
Further, 6-chlorohexanal (12) could be obtained quite conveniently from compound 18 in two steps by employing SOCl2 and HCOOH and was isolated in a total yield of 85%. Compound 3 could be obtained via a Fischer reaction between compound 11 and aldehyde 12. For the purpose of increasing the yield of compound 3, a variety of parameters such as the solvent, catalyst, temperature, and reaction time were screened to determine the optimum reaction condition, and the results are summarized in Table 1. The experimental results indicate that raising the reaction temperature to 170°C led to a decrease in the yield, accompanied with the formation of additional by-product, as detected by thin-layer chromatography (TLC). Following this screening, it was concluded that EtOH/H2O and 20% H2SO4 was a better system to accomplish this Fischer transformation because of the better yield (70%) and lower cost.9–12
Optimization of the Fischer indole reaction in the synthesis of compound 3.
Entry
Solvent
Catalyst
Temperature (°C)
Time (h)
Yield (%)
1
EtOH/H2O
20% CH3COOH
80
4
–
2
EtOH/H2O
36.5% HCl
80
4
35
3
EtOH/H2O
20% H2SO4
80
4
70
4
EtOH/H2O
10% TFA
80
4
68
5
n-butanol/H2O
20% CH3COOH
110
4
–
6
n-butanol/H2O
36.5% HCl
110
4
34
7
n-butanol/H2O
20% H2SO4
110
4
65
8
n-butanol/H2O
10% TFA
110
4
68
9
Ethylene glycol/H2O
20% CH3COOH
170
2
–
10
Ethylene glycol/H2O
36.5% HCl
170
2
23
11
Ethylene glycol/H2O
20% H2SO4
170
2
47
12
Ethylene glycol/H2O
10% TFA
170
2
51
With compound 3 in hand, we next attempted to explore an efficient route to synthesize carboxamide 6 by using 5-bromo-2-hydroxybenzaldehyde (16) as the starting material (Scheme 6). Initially, acid 21 was easily prepared via condensation between compound 16 and ethyl 2-bromoacetate and was isolated in an excellent yield (90%).13,14 The piperazine moiety can be easily introduced through a C–N cross-coupling reaction under basic conditions to give the intermediate 22. Although the yield of 22 was not satisfactory, we found that any unconverted compound 21 could be easily recycled. After the reaction, HCl was added to the reaction solution, and the unreacted compound 21 was salted out, while the intermediate 22 stayed in the reaction solution. Finally, intermediate 22 was transformed into carboxamide 6 in 80% yield by treatment with SOCl2 and NH3 (two steps without isolation).15,16
The efficient synthesis of key intermediate 6.
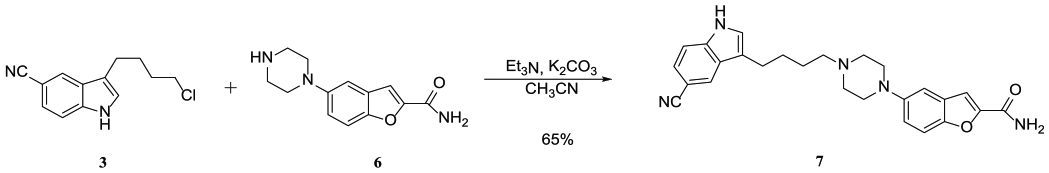
Subsequently, vilazodone (7) was prepared via nucleophilic substitution between chloride 3 and carboxamide 6 (Scheme 7). The experimental results indicated that the Et3N/K2CO3 system was very effective for this transformation, and the desired product vilazodone (7) was obtained in 65% yield.1
Synthesis of vilazodone (7).
Conclusion
In conclusion, we have developed an efficient and potentially commercial process for the synthesis of vilazodone (7) by employing 4-aminobenzonitrile and 5-bromo-2-hydroxybenzaldehyde as starting materials. This novel synthetic route involves diazotization, a Fischer reaction, cyclization, and substitution to afford vilazodone in an overall yield of 24% and with 99% purity. It is noteworthy that the reagents and materials used in this synthetic route are inexpensive, and the process should be suitable for industrial production.
Experimental
Melting points were determined using a Buchi digital melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Varian spectrometer at working frequencies of 400 and 100 MHz, respectively, in DMSO-d6 using tetramethylsilane (TMS) as an internal standard. All chemical shifts are reported as δ values (ppm) relative to TMS and observed coupling constants (J) are given in Hertz (Hz). MS data were obtained on an instrument of Agilent 6545 Q-TOF. All reagents were purchased from commercial sources and used without prior purification. Column chromatography was performed on silica gel (200–300 mesh) eluting with n-hexane/ethyl acetate.
4-Hydrazinylbenzonitrile hydrochloride (11): 4-Aminobenzonitrile (118 g, 1.0 mol) was dissolved in hydrochloric acid (36.5%, 300 g, 3.0 mol in 500 mL of H2O). NaNO2 (79.5 g, 1.1 mol) and H2O (200 mL) were added into the solution slowly for 1 h at 2°C. Then, reaction mixture was filtered, and the residue was dried for 12 h and identified as by-product 20 (12.3 g). The filtrate was transferred to a new vessel and NaHSO3 (520 g, 5.0 mol) was added, and the mixture was stirred at 85°C for 2 h. After filtering, the filtrate was distilled to remove one-third of the solvent, and then, 36.5% HCl (1 L) was added to the residue solution. After stirring at 50°C for 30 min, the mixture was filtered, and the product was obtained as a white solid. Yield 137 g (81%). High-performance liquid chromatography (HPLC) purity 94.2%. m.p. 238.8–240.5°C. (Lit17 240°C). 1H NMR (400 MHz, D2O): δ 7.54–7.32 (m, 2H), 6.93–6.83 (m, 2H), 4.67 (s, 1H), 3.33 (s, 2H). 13C NMR (100 MHz, D2O): δ 158.7, 134.6, 134.6, 122.3, 114.5, 114.5, 103.3. HRMS (ESI): m/z [M + H]+ calculated for C7H8ClN3: 170.0480, found: 170.0477.
3-(4-Chlorobutyl)-1H-indole-5-carbonitrile (3): Compound 11 (169 g, 1 mol) was dissolved in EtOH/H2O (1:1, 100 mL), and then, 20% H2SO4 (20 mL) and 6-chlorohexanal 12 (134 g, 1.0 mol) were added. The mixture was stirred at 80°C for 4 h. After the reaction was complete, the aqueous was extracted with CH2Cl2. The combined CH2Cl2 fractions were evaporated to afford product 3 as a milky-white solid. Yield 162 g (70%), 97% purity. m.p. 97.6–99.5°C. (Lit1 99–99.5°C) 1H NMR (400 MHz, DMSO-d6): δ 11.38 (s, 1H), 8.08 (s, 1H), 7.55–7.25 (m, 3H), 3.67 (t, J = 6.1 Hz, 2H), 2.75 (t, J = 6.7 Hz, 2H), 1.77 (dd, J = 6.3, 3.1 Hz, 4H). 13C NMR (100 MHz, DMSO-d6): δ 137.8, 127.2, 124.8, 124.5, 123.2, 120.7, 117.1, 111.9, 102.2, 44.9, 32.4, 27.3, 24.2. HRMS (ESI): m/z [M + Na]+ calculated for C13H13ClN2: 255.0659, found: 255.0657.
5-Bromobenzofuran-2-carboxylic acid (21): 5-Bromo-2-hydroxybenzaldehyde (16) (201 g, 1.0 mol) was dissolved in ethyl acetate (1 L), and then, BrCH2COOCH2CH3 (334 g, 2 mol) and K2CO3 (410 g, 3 mol) were added. The mixture was stirred and refluxed for 6 h. After the reaction was complete, the liquid was filtered and the filtrate was concentrated under vacuum. KOH (84 g, 1.5 mol) and H2O (500 mL) were added to the residue, and the resulting mixture was stirred at 80°C for 3 h. Subsequently, HCl (3 mol L−1) was added to adjust the pH to 2. After filtration, the product 21 was obtained as a white solid. Yield 217 g (90%) with 94% purity. m.p. 255.5–256.8°C. 1H NMR (400 MHz, DMSO-d6): δ 13.64 (s, 1H), 7.95 (d, J = 1.9 Hz, 1H), 7.77–7.46 (m, 3H). 13C NMR (100 MHz, DMSO-d6): δ 159.4, 153.0, 146.9, 129.9, 128.8, 125.1, 115.8, 114.0, 112.5. HRMS (ESI): m/z [M + H]+ calculated for C9H5BrO3: 240.9495, found: 240.9490.
5-(Piperazin-1-yl)benzofuran-2-carboxylic acid (22): Compound 21 (241 g, 1.0 mol) and piperazine (430 g, 5 mol) were dissolved in dimethylformamide (DMF), and the mixture was stirred at 140°C for 6 h. After the reaction was complete, HCl (10 mol L−1, 500 mL) was added. After filtration, KOH/H2O was added into the filtrate until pH 6–7. The solution was extracted with CH2Cl2, and then, the organic phase was concentrated under vacuum to obtain product 22 as a faint yellow solid. Yield 221 g (90%) with 95% purity. m.p. 275–278.2°C. (Lit18 274–276°C). 1H NMR (400 MHz, DMSO-d6): δ 10.58 (s, 1H), 8.19–7.28 (m, 4H), 3.02 (d, J = 4.2 Hz, 4H), 2.86 (s, 4H), 2.51 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 159.6, 149.6, 148.2, 126.8, 117.9, 110.9, 109.6, 107.7, 107.3, 52.3(2), 45.5(2). HRMS (ESI): m/z [M + H]+ calculated for C13H14N2O3: 247.1077, found: 247.1080.
5-(Piperazin-1-yl)benzofuran-2-carboxamide (6): Compound 22 (246 g, 1.0 mol) was dissolved in ClCH2CH2Cl, and SOCl2 (119 g, 1 mol) was added slowly to the solution and the resulting mixture was stirred at 60°C for 4 h. After filtration, the solid was dissolved in tetrahydrofuran (THF) (1 L), and then, NH3 gas (0.3 MPa in high-pressure reactor) was poured into the solution. The resulting mixture was stirred at 25°C for 30 min. After the reaction was complete, the mixture was concentrated under vacuum to provide the product 6. Yield 196 g (80%) with 97% purity. m.p. 251.3–254.3°C. (Lit19 253–255°C). 1H NMR (400 MHz, DMSO-d6): δ 8.19–7.28 (m, 4H), 7.15 (d, J = 6.6 Hz, 2H), 3.02 (d, J = 4.2 Hz, 4H), 2.86 (s, 4H), 2.51 (s, 1H). 13C NMR (100 MHz, DMSO-d6): δ 159.9, 149.2, 148.6, 127.5, 117.9, 111.4, 109.6, 107.3, 107.2, 51.0(2), 45.71, 45.7. HRMS (ESI): m/z [M + H]+ calculated for C13H15N3O2: 246.1237, found: 246.1245.
Vilazodone (7): Compound 3 (233 g, 1.0 mol) and 6 (245 g, 1.0 mol) were dissolved in acetonitrile (3 L), and then, triethylamine (101 g, 1.0 mol) and K2CO3 (138 g, 1.0 mol) were added to the solution. The mixture was heated at reflux for 12 h. Subsequently, the reaction mixture was poured into cold water (3 L). After filtration, the solid was dissolved in EtOAc, and then, HCl-EtOAc saturated solution was added to enable crystallization. Product 7 was obtained as a white solid. Yield 287 g (65%) with 99% HPLC purity after the mixture was filtered. m.p. 276.5–279.2°C (Lit1 277–279°C). 1H NMR (400 MHz, DMSO-d6): δ 11.35 (s, 1H), 8.04 (d, J = 18.3 Hz, 2H), 7.74–6.88 (m, 8H), 3.69–3.15 (m, 8H), 2.74 (t, J = 7.2 Hz, 2H), 2.37 (t, J = 7.2 Hz, 2H), 1.80–1.61 (m, 2H), 1.54 (d, J = 6.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 159.9, 148.8, 147.9, 138.6, 137.7, 127.8, 126.9, 124.7, 124.0, 123.3, 120.9, 117.8, 115.7, 112.4, 111.6, 109.5, 107.3, 100.0, 57.5, 52.8(2), 49.8(2), 27.7, 26.1, 24.1. HRMS (ESI): m/z [M + H]+ calculated for C26H27N5O2: 442.2238, found: 442.2240.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (21406200) and the Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals.
ORCID iD
Weike Su
References
1.
HeinrichTBöttcherHGerickeR, et al. J Med Chem2004; 47: 4684–4692.
2.
HeinrichTBöttcherH.Bioorg Med Chem Lett2004; 14: 2681–2684.
3.
HuBSongQXuY.Org Process Res Dev2012; 16: 1552−1557.