5-Carbohydrazides and 5-carbonylazides of pyrazolo[3,4-b]pyridines are used to synthesize new heterocyclic derivatives. Some unexpected behaviors are observed in the reactions of the above two species. The structures of the obtained compounds are proved by spectroscopic studies together with elemental and X-ray structure analyses.
The pyrazolo[3,4-b]pyridine ring system has emerged as an important and pharmaceutically relevant scaffold in view of its occurrence as part of bioactive drugs and numerous biologically active compounds.1 These compounds show a wide spectrum of biological diversity, such as antiviral,2 anti-inflammatory,3 anxiolytic,4 hypoglycemic,5 antitumor,6 herbicidal,7 antiherpetic, and antiallergic.8 They also act as serotonin re-uptake inhibitors,9 cholecystokinin (CCK) agonists,10 vasodilators,11 potent cyclin-dependent kinase 1 inhibitors,12 HIV reverse transcriptase inhibitors,13 CCR1 antagonists,14 and protein kinase inhibitors.15 Some pyrazolo[3,4-b]pyridine-embedded heterocycles are anxiolytic drugs16 such as cartazolate, etazolate, and tracazolate. They are also present in the cardiovascular therapeutic agent, BAY 41-2272,17 and in a glycogen synthase kinase 3 (GSK-3) inhibitor that is efficacious in the treatment of Alzheimer’s disease.18 In addition to their biological importance, their structural similarity to purine, an important constituent of DNA and RNA nucleosides,18 is an added attraction that increases interest in the synthesis of pyrazolo[3,4-b]pyridines. Recently, we found that heating 3-methyl-6-oxo-1-phenyl-6,7-dihydro-1H-pyrazolo[34-b]pyridine-5-carbonylazide (1) in MeOH resulted in a Curtius rearrangement to give 3-methyl-1-phenyl-1,5-dihydro-6H-oxazolo[5,4-b]pyrazolo[4,3-e]-pyridin-6-one (2) (Scheme 1).19
Conversion of compound 1 via Curtius rearrangement into oxazolo[5,4-b]pyrazolo[4,3-e]pyridin-6-one 2.
A series of pyrazolopyridine derivatives was described by Geraldo et al.20 as potential antithrombotic lead molecules due to their optimal inhibitory activity against platelet aggregation induced by collagen and arachidonic acid, both potent physiological stimuli for thrombus formation.20 In continuation of our work on pyrazolo[3,4-b]pyridines,19,21–25 we aimed to synthesize various derivatives of these compounds, including heterocyclic systems from reactions of 3,6-dimethyl-1-phenyl-1H-pyrazolo[3,4-b]pyridine-5-carbohydrazide (4) (see Scheme 2) and 3-methyl-6-oxo-1-phenyl-6,7-dihydro-1H-pyrazolo-[3,4-b]pyridine-5-carbohydrazide (15) (see Scheme 4) with various reagents via functional group manipulation. One of these reactions involved the Curtius rearrangement, which has been widely used in organic synthesis due to the utility of isocyanate intermediates.26
Synthesis and reactions of various pyrazolo[3,4-b]pyridine-5-carbohydrazides 4. Reagents and conditions: (i) NH2NH2·H2O, reflux 5 h; (ii) PhNCS (5a) or CH2=CHCH2NCS (5b), EtOH, reflux 8–10 h; (iii) EtOH, piperidine, reflux 8–10 h; (iv) NaNO2/HCl, 0–5 °C; (v) xylene/piperidine, reflux 12 h; (vi) toluene, reflux 8 h.
Results and discussion
Previously, it was reported that 3,6-dimethyl-1-phenyl-1H-pyrazolo[3,4-b]pyridine-5-carbohydrazide (4) was synthesized by the reaction of pyrazolo[3,4-b]pyridine-5-carboxylate [19] with an excess amount of hydrazine hydrate for 5 h (Scheme 2). In this context, we describe the reaction of compound 4 with various reagents such as phenyl isothiocyanate in order to synthesize a range of heterocyclic derivatives. In refluxing EtOH, the reaction between 4 and phenyl isothiocyanate (5a) afforded, after 8 h, 2-(3,6-dimethyl-1-phenyl-1H-pyrazolo[3,4-b]pyridine-5-carbonyl)-N-phenyl-hydrazine-1-carbothioamide (6a) in 89% yield (Scheme 2).
In the case of the reaction between 4 and allyl isothiocyanate (5b), the reaction proceeded after 10 h in refluxing EtOH to give compound 6b in 70% yield (Scheme 2). The 1H NMR spectrum of 6b revealed three singlets due to NH protons at δ 10.57, 8.50, and 8.12 related to hydrazine-NH-thiourea, hydrazino-amide, and NH-CH2-, respectively. The allyl protons appeared as three multiplets at δ 4.21–4.26 (allyl CH2N), 5.07–5.16 (allyl CH2=), and 5.95–6.01 (allyl CH=). The 13C NMR spectrum of 6b also supported the assigned structure with the appearance of allyl carbon signals at δ 44.2 (allyl CH2N), 105.8 (allyl CH2=), and 129.3 (allyl CH=). Besides, the thioamide and carbonyl carbon signals appeared at δ 178.0 and 164.4, respectively.
On reacting 4 with various carbonyl compounds 7a–f in EtOH and catalyzed by piperidine, the corresponding condensed products 8a–f were obtained (Scheme 2). The distinctive carbons of compound 8b are shown in Figure 1, whereas the spectroscopic data of compound 8b are listed in Table 1. In the case of 8f, the 1H NMR spectrum showed five singlets at δ 13.30, 11.20, 8.32, 2.80, and 2.50 related to hydrazine-NH, istatin-NH, pyridine-H-4, pyridine-CH3, and pyrazole-CH3, respectively. In 13C NMR spectrum of 8f, five distinctive carbon signals resonated at δ 167.9, 165.9, 131.9, 23.9, and 12.2 due to isatin-C=O, hydrazine-CO, pyridine-H, pyridine-CH3, and pyrazole-CH3, respectively.
COSY: correlation spectroscopy; HSQC: heteonuclear multiple bond correlation; HMBC: heteronuclear multiple bond correlation
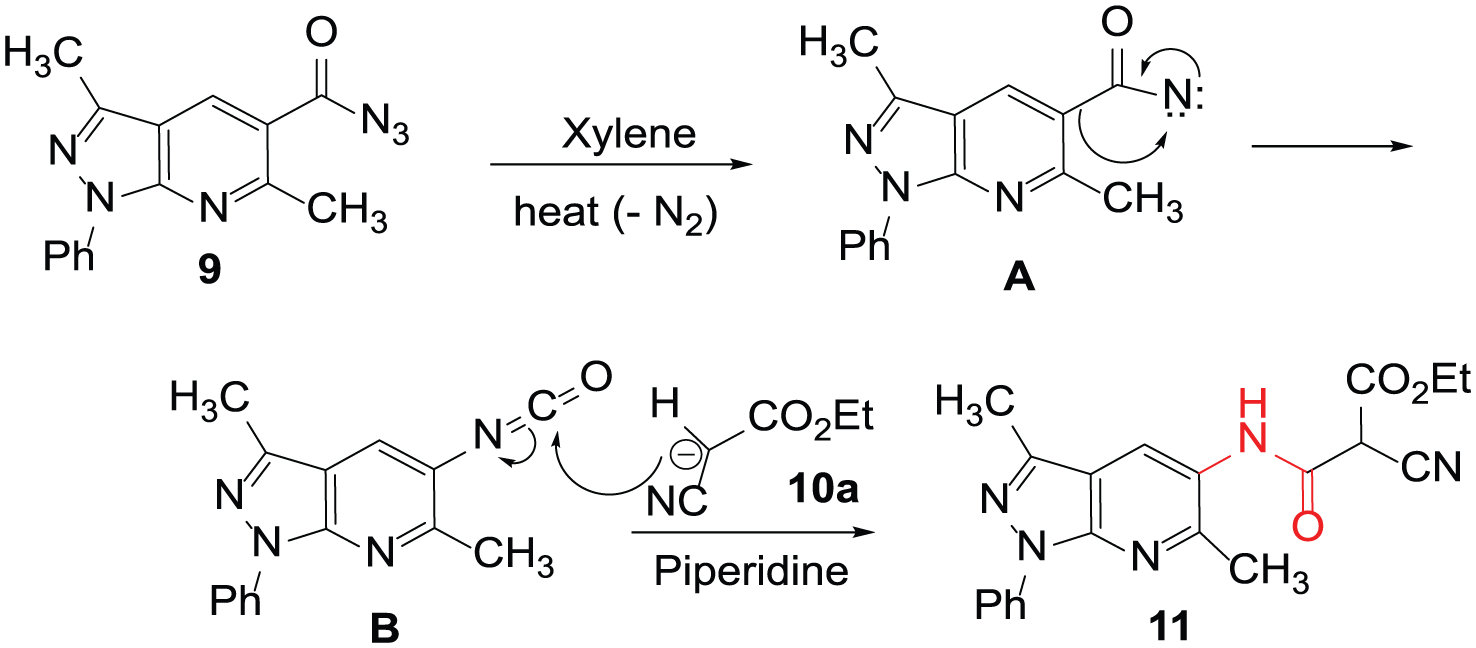
Interestingly, the reaction of 4 with NaNO2/HCl led to the formation of compound 919 in 75% yield (Scheme 2). On subjecting compound 9 to react with ethyl 2-cyanoacetate (10a) in xylene as the solvent and catalyzed by a few drops of piperidine, the new cyanoester 11 was obtained in 65% yield (Scheme 2). The elemental analysis and mass spectrum of compound 11 were in agreement with its molecular formula (C20H19N5O3). The ester protons of 11 appeared in the 1H NMR spectrum as a triplet (3H) and a quartet (CH2) at δ 1.49 and 3.47, respectively. The carbonyl-ester and amide carbons resonated at δ 164.5 and 163.9, whereas the methyl carbon signals appeared at δ 12.20, 18.9, and 22.4 for the methyl-ester, methyl-pyrazole, and methyl-pyridine, respectively. The proton of the tert-carbon (CH) appeared at δ 4.10 in the 1H NMR spectrum, whereas the corresponding carbon signal appeared at δ 46.8.
The mechanistic explanation is based upon extrusion of N2 from 9 under heating to form intermediate A (Figure 2). Rearrangement of A via a Curtius pathway would form isocyanate B. Nucleophilic addition of the formed anion, from the deprotonation of 10a by piperidine, to the carbonyl of the isocyanate would form compound 11 (Figure 2).
Mechanism for the formation of 11 from the reaction of 9 with 10a.
The reaction of 4 with diethyl acetylenedicarboxylate (12) in refluxing toluene gave the corresponding pyrazolone derivative 13 in 90% yield (Scheme 2). The 1H NMR of 13 showed a signal at δ 11.20 for the pyrazolone-NH proton. The peak at δ 8.20 corresponded to the pyridine-H-4 proton. The pyrazolone structure was proved by the appearance of the pyrazolone-H resonating at δ 6.82, whereas the ester protons resonated at δ 1.30 as a triplet (J = 7.0 Hz) and at δ 3.75 as a quartet (J = 7.0 Hz). The ester carbon signals appeared in the 13C NMR spectrum at δ 15.7 (CH3) and 52.8 (CH2). In addition, the pyrazolone-CH carbon appeared at δ 124.6, and the carbonyl-pyrazolone resonated at δ 163.3 (see the experimental analysis section).
Similarly, treatment of compounds 6a,b with 12 afforded the thiazolone derivatives 14a,b (Scheme 3). For products 14a and 14b, the 1H NMR and 13C NMR spectra were found to be in accordance with their proposed structures. For example, the 13C NMR spectrum of 14a showed three carbon signals at δ 167.0, 165.9, and 163.0 for the carbonyl-carbons of the ester, thiazolone, and carbohydrazine, respectively. In addition, three carbon signals for three C=N carbons appeared at δ 157.2, 152.6, 150.0 (see the experimental analysis section). The ester carbons appeared at δ 60.8 (CH2) and δ 11.6 (CH3). In the case of 14b, the 13C NMR spectrum revealed the exocyclic thiazolone-CH at δ 128.0, whereas the allyl carbons appeared at δ 43.4 (allyl CH2N), 103.8 (allyl CH2=), and 132.1 (allyl CH=).
Synthesis of 3,6-dimethyl-thiazolopyrazolo[3,4-b]pyridines 14a,b.
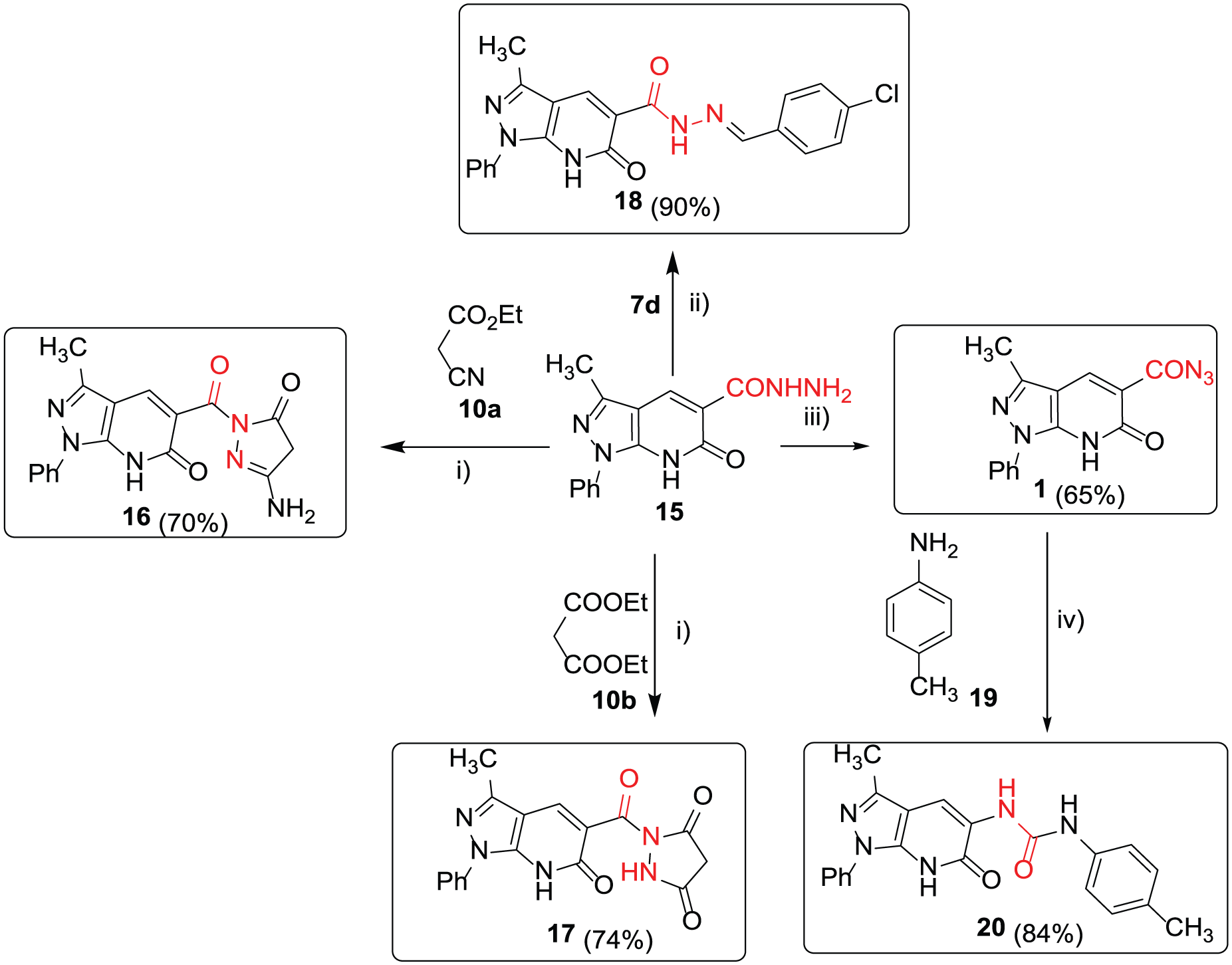
On reacting the second class of carbonylhydrazide derivative of pyrazolo[3,4-b]pyridone 15 [19] with active methylenes such as ethyl 2-cyanoacetate (10a) and/or diethyl malonate (10b) in AcOH acid and catalyzed by a few drops of conc. H2SO4, the reaction produced compounds 16 and 17 (Scheme 4).
Cyclo-condensation and rearrangement reactions of 2-oxo-pyrazolo[3,4-b]-pyridine-carbonylhydrazide 15. Reagents and conditions: (i) AcOH/H2SO4, reflux, 10 h; (ii) 4-Cl-C6H4-CHO, EtOH/piperidine, reflux, 8 h; (iii) NaNO2/HCl, 0–5 °C; (iv) xylene/piperidine, reflux, 6 h.
The 1H NMR spectrum of 16 showed the pyridone-NH, pyridone-CH-4, and NH2 protons as three singlets at δ 10.55 (1H), 8.65 (1H), and 6.30 (2H), respectively. Besides, two other singlets appeared at δ 2.70 and 2.00, corresponding to pyrazole-CH2-4 and pyrazole-CH3 (see the experimental analysis section). The 13C NMR spectrum of 16 showed three distinctive carbonyl carbon signals of the pyrazolone at δ 166.8, the carbonylhydrazine at δ 163.4, and the carbonyl-2-pyridine at δ 162.3. Most indicative is the presence of the 4H-pyrazolone at δ 2.70 in the 1H NMR spectrum and at δ 66.3 in the 13C NMR spectrum. In the case of 17, five singlets apparent in the 1H NMR spectrum at δ 10.98 (1H), 9.30 (1H), 8.65 (1H), 3.50 (3H), and 2.45 (3H) were corresponding to the pyridone-NH, pyrazole-NH, pyridone-H-4, pyridone-CH3, and pyrazole-CH3, respectively. In the 13C NMR spectrum, the carbonyl carbon signals resonated at δ 166.8 (ester), 163.4, 162.3 (pyrazoledione), and 160.0 (pyridone-CH-4).
Reaction of equimolar amounts of the carbohydrazide 15 and p-chlorobenzaldehyde (7d) in refluxing EtOH gave yellow crystals of compound 18 in 90% yield. The structure of 18 was proved by nuclear magnetic resonance (NMR) spectroscopy. Five singlets appeared at δ 12.00 (NH-pyridone), 9.00 (carbonylhydrazine), 8.60 (azomethine), 8.15 (pyridinone-H-4), and 2.50 (CH3) in the 1H NMR spectrum. The 13C NMR spectrum of 18 revealed the two carbonyl carbon signals at δ 165.3 and 163.6. The two azomethine carbon signals appeared at δ 156.9 and 147.6. The E form of compound 18 was proved via X-ray structure analysis (Figure 3).
Molecular structure of one of the crystallographic independent molecules of 18. (E)-N’-(4-Chlorobenzylidene)-3-methyl-6-oxo-1-phenyl-3a,6,7,7a-tetrahydro-1H-pyrazolo [3,4-b]pyridine-5-carbohydrazide (displacement parameters are drawn at the 50% probability level).
Treatment of 1519 with NaNO2/HCl at 0–5 °C led to the formation of 119 (Scheme 4). Treatment of 1 with p-toluidine (19) occurred with thermolysis and extrusion of N2, accompanied by rearrangement to give urea derivative 20 in 84% yield (Scheme 4). The structure of 20 was confirmed by the presence of six singlets resonating in the 1H NMR spectrum at δ 10.70 (1H, NH), 9.85 (1H, NH), 8.55 (1H, pyridone-H), 8.25 (1H, NH), 3.35 (3H, CH3), and 2.35 (3H, CH3).
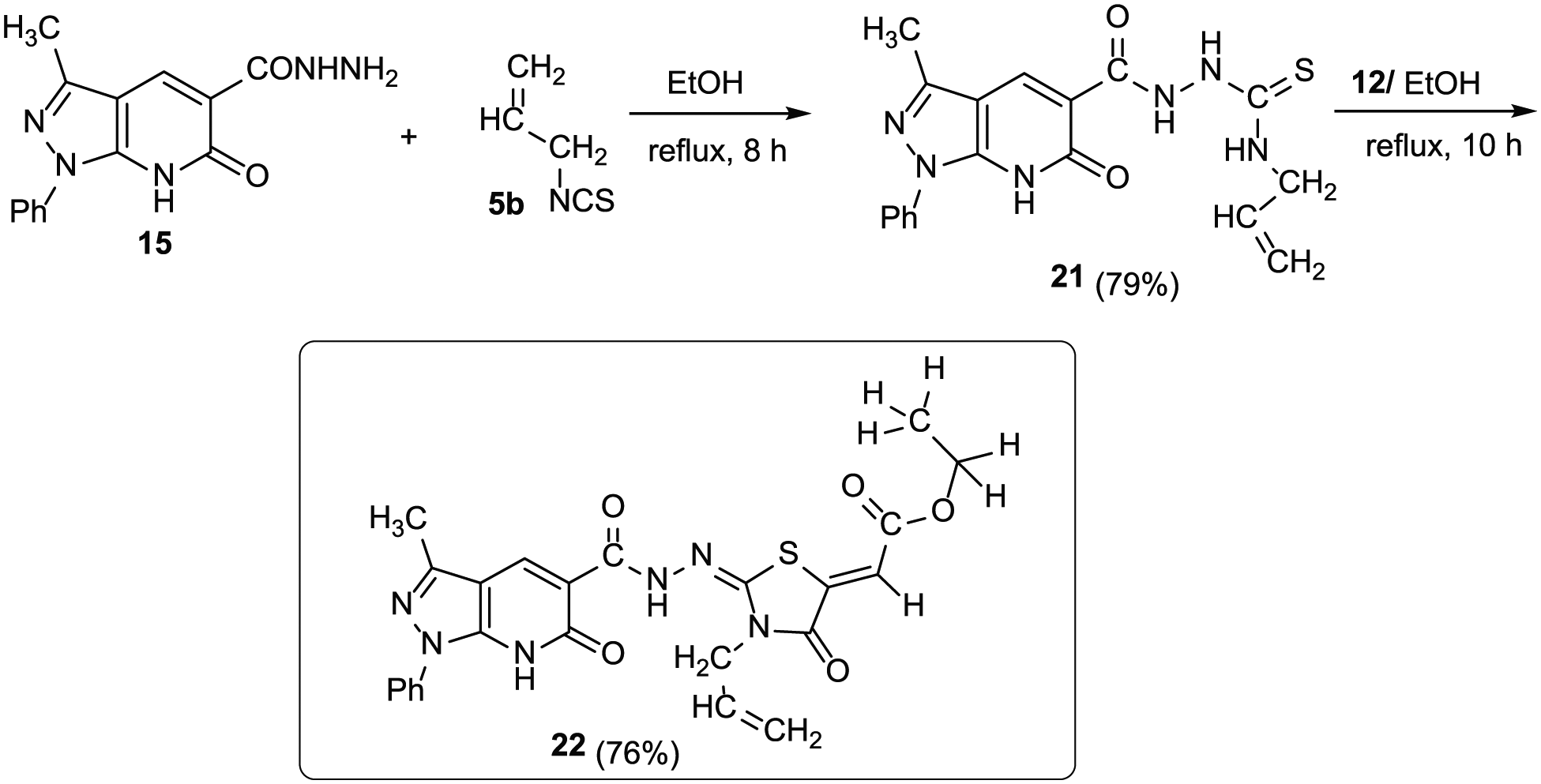
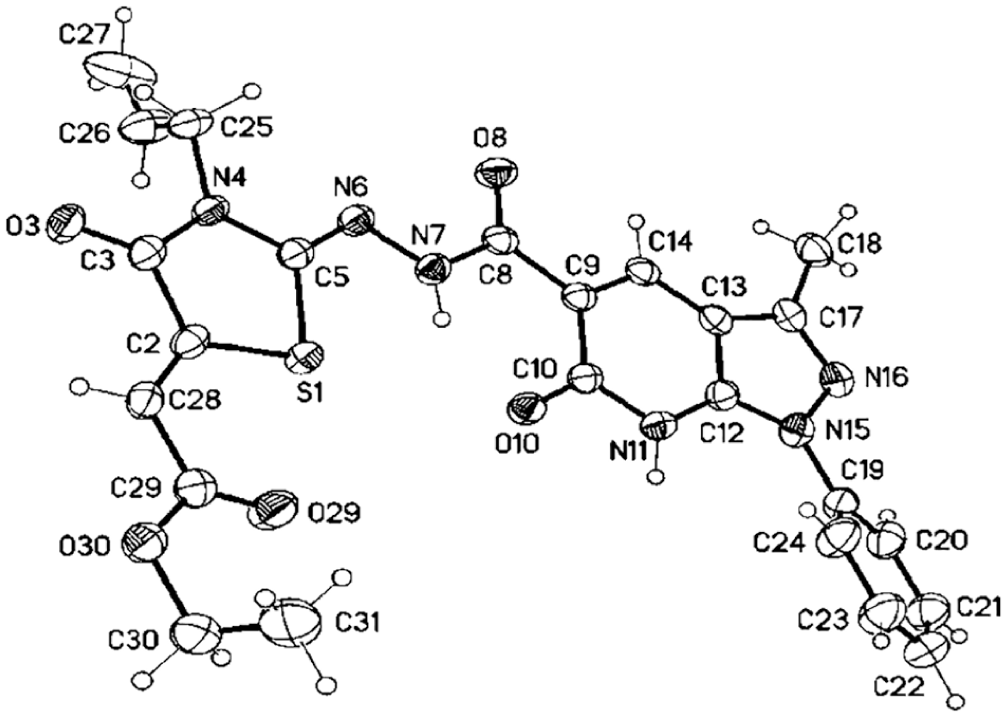
For further investigation of the reaction of the carbohydrazine group in compound 15, we reacted it with allyl isothiocyanate (5b) in refluxing EtOH, and N-allyl-2-(3-methyl-6-oxo-1-phenyl-6,7-dihydro-1H-pyrazolo[3,4-b]pyridine-5-carbonyl)hydrazine-1-carbothioamide (21) was obtained in 79% yield (Scheme 5). The 1H NMR spectrum of 21 revealed the four NH protons at δ 11.20, 9.90, 9.20, and 7.50 (see the experimental analysis section). Three multiplets were also observed at δ 4.26–4.31, 5.12–5.18, and 5.90–6.94 related to the allyl CH2N, allyl CH2=, and allyl-CH2 moieties, respectively. The allyl carbons resonated in the 13C NMR spectrum at δ 46.5, 106.4, and 134.0. Moreover, the carbothioamide-C, pyridone-C, and carbonyl-hydrazine-C resonated at δ 178.2, 166.2, and 163.4, respectively. Subsequently, reaction of 21 with 12 in EtOH afforded the corresponding pyrazolo[3,4-b]pyridine thiazole derivative 22 in 76% yield (Scheme 5). The 1H NMR spectrum revealed the NH protons at δ 11.15 and 9.25 assignable to the pyridone-NH and amide-NH, whereas the other NH protons had disappeared, indicating that they were involved in the cyclization process. The ester carbon signals appeared at δ 168.7, 57.0, and 13.5 for the carbonyl-ester, CH2-ester, and CH3-ester, respectively. Besides, the exocyclic vinyl thiazolidine and the two C=N carbons appeared at δ 135.6, 154.6, and 153.3, respectively. The structure of compound 22 was finally confirmed by X-ray structure analysis (Figure 4).
Synthesis of 3-methyl-thiazolopyrazolo[3,4-b]pyridin-6-one 22.
Molecular structure of one of the crystallographic independent molecules of 22. Ethyl (Z)-2-((Z)-3-allyl-2-(2-(3-methyl-6-oxo-1-phenyl-6,7-dihydro-1H-pyrazolo[3,4-b]pyridine-5-carbonyl)-hydrazineylidene)-4-oxothiazolidin-5-ylidene)acetate (displacement parameters are drawn at the 50% probability level).
Conclusion
In conclusion, we have described the utility of carbohydrazides of pyrazolo[3,4-b]pyridine as versatile precursors to synthesize various new heterocyclic derivatives, which might be important in different biological and pharmaceutical contexts.
Experimental analysis
General
Melting points were determined using an APP Digital ST 15 melting point apparatus and are uncorrected. Thin-layer chromatography (TLC) analyses were performed using analytical Merck 9385 silica aluminum sheets (Kieselgel 60) with PF254 indicator. The infrared (IR) spectra were recorded as KBr disks on a Shimadzu-408 infrared spectrophotometer, at the Faculty of Science, Minia University. The NMR spectra were measured using a Bruker AV-400 spectrometer at the Karlsruhe Institut für Technologie (KIT), Institute of Organic Chemistry, Karlsruhe, Germany. Chemical shifts are expressed as δ (ppm) with tetramethylsilane as an internal reference. The samples were dissolved in dimethyl sulfoxide-d6 (DMSO-d6), where s = singlet, d = doublet, dd = doublet of doublets, and t = triplet. Mass spectra were recorded on a Varian MAT 312 instrument in EI mode (70 eV), at the KIT, Institute of Organic Chemistry, Karlsruhe, Germany. Elemental analyses were obtained using a Varian Elementary device at the National Research Center, Giza, Egypt.
Starting materials
Compounds 1, 3, 4, 9, 12, and 15 were prepared according to literature procedures [19].
Crystal structure determinations of 18 and 22
The single-crystal X-ray diffraction studies were carried out on a Bruker D8 Venture diffractometer with a PhotonII CPAD detector at 123(2) K using Cu-Kα radiation (λ = 1.54178 Å). Dual space methods (SHELXT)27 were used for structure solution, and refinement was carried out using SHELXL-2014 (full-matrix least-squares on F2).28 Hydrogen atoms were refined using a riding model (H(N, O) free). Semi-empirical absorption corrections were applied. In 18, refinement with the listed atoms shows residual electron density due to two heavily disordered EtOH solvent molecules in the unit cell, which could not be refined with a split atom model. Therefore, the option “SQUEEZE” of the program package PLATON29 was used to create a hkl file taking into account the residual electron density in the void areas (see cif-file for details).
18: Yellow crystals, C21H16ClN5O2∙2 H2O·½ C2H6O, Mr = 464.90, crystal size 0.24 × 0.04 × 0.02 mm3, triclinic, space group P-1 (No. 2), a = 7.7981(2) Å, b = 17.1490(5) Å, c = 18.6509(5) Å, α = 62.718(2)°, β = 82.813(2)°, γ = 89.055(2)°, V = 2196.95(11) Å3, Z = 4, ρ = 1.406 Mg/m−3, µ(Cu-Kα) = 1.906 mm−1, F(000) = 972, 2θmax = 145.2°, 30,237 reflections, of which 8623 were independent (Rint = 0.057), 597 parameters, 16 restraints, R1 = 0.048 (for 6556 I > 2σ(I)), wR2 = 0.123 (all data), S = 1.03, largest diff. peak/hole = 0.832/−0.302 e Å−3.
22: Yellow crystals, C24H22N6O5S·2 H2O, Mr = 542.57, crystal size 0.20 × 0.06 × 0.03 mm3, monoclinic, space group P21/n (No. 14), a = 9.9686(4) Å, b = 22.6653(10) Å, c = 22.4652(10) Å, β = 97.078(2)°, V = 5037.1(4) Å3, Z = 8, ρ = 1.431 Mg/m−3, µ(Cu-Kα) = 1.638 mm−1, F(000) = 2272, 2θmax = 144.2°, 55,336 reflections, of which 9920 were independent (Rint = 0.042), 723 parameters, 16 restraints, R1 = 0.067 (for 8138 I > 2σ(I)), wR2 = 0.180 (all data), S = 1.10, largest diff. peak/hole = 0.674/−0.352 e Å−3.
CCDC 1905952 (18) and CCDC 1905953 (22) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
Preparation of compounds 6a,b
A mixture of compound 4 (0.50 g, 1.77 mmol) and 5a (0.24 g, 1.77 mmol) or 5b (0.18 g, 1.77 mmol) was heated under reflux for 8–10 h in EtOH (100 mL). The solution was cooled and poured onto ice cold water, and the obtained products 6a,b were filtered off, washed with water, dried, and were recrystallized from the stated solvents.
A mixture of compound 4 (0.50 g, 1.8 mmol) and carbonyl compounds 7a–f (1.8 mmol) was heated under reflux in EtOH (50 mL) and a few drops of piperidine for 8–10 h. The reaction mixture was poured onto ice cold water (200 mL), and the products 8a–f were filtered off, washed with water, and recrystallized from the stated solvents.
A mixture of compound 9 (0.50 g, 1.7 mmol) and ethyl cyanoacetate 10a (0.19 g, 1.7 mmol) in xylene (50 mL) containing piperidine (0.3 mL) was heated under reflux for 12 h. The solution was cooled and poured onto petroleum ether (100 mL), and the product 11 was filtered off, washed with petroleum ether, and dried.
Reaction of 4 with diethyl acetylenedicarboxylate (12)
A mixture of 4 (2.81 g, 10 mmol) with 12 (1.7 g, 10 mmol) in toluene was heated under reflux for 8 h. The solution was cooled and poured onto petroleum ether (60/80) (100 mL), and the product 13 was then filtered off, washed with petroleum ether, and dried.
Reaction of 6a,b with 12; Synthesis of compounds 14a,b
A mixture of 6a or 6b (10 mmol) with 12 (1.7 g, 10 mmol) in toluene (100 mL) was heated under reflux for 8–10 h. The solution was cooled, and the solid products 14a,b were filtered off, washed with water, dried, and recrystallized from the stated solvents.
A mixture of the carbohydrazide 15 (0.50 g, 1.8 mmol) and 10a (0.2 g, 1.8 mmol) or 10b (0.28 g, 1.8 mmol) was heated under reflux for 10–12 h in acetic acid (20 mL) and a few drops of sulfuric acid. The solution was cooled and poured onto ice-cold water (100 mL). The products 16 or 17 were filtered off, washed with water, dried, and crystallized from the stated solvents.
A mixture of the carbohydrazide 15 (0.50 g, 1.8 mmol) and 4-chlorobenzaldehyde 7d (0.25 g, 1.8 mmol) was heated under gentle reflux for 10 h in EtOH and a few drops of piperidine. The solution was cooled and poured onto ice cold water (100 mL), and the product 18 was filtered off, washed with water (100 mL), and dried.
A mixture of compound 1 (0.50 g, 1.7 mmol) and p-toluidine 19 (0.18 g, 1.8 mmol) was heated under reflux for 6 h in xylene (50 mL) and piperidine (0.2 mL). The solution was cooled and poured onto petroleum ether (60/80 °C) (100 mL), and the product 20 was filtered off and dried.
A mixture of compound 15 (2.83 g, 10 mmol) and 5b (1.7 g, 10 mmol) was heated under reflux for 8 h in EtOH (20 mL). The solution was cooled and poured onto water, and the product 21 was filtered off and dried.
A mixture of compound 21 (3.82 g, 10 mmol) and 12 (1.7 g, 10 mmol) was heated under reflux for 10 h in EtOH (100 mL). The solution was cooled and poured onto water, and the formed product 22 was filtered off and dried.
ed_ashraf-talaat_24-1-2019_Suppl_Data_of_second_paper – Supplemental material for 5-Carbohydrazide and 5-carbonylazide of pyrazolo[3,4-b]pyridines as reactive intermediates in the synthesis of various heterocyclic derivatives
Supplemental material, ed_ashraf-talaat_24-1-2019_Suppl_Data_of_second_paper for 5-Carbohydrazide and 5-carbonylazide of pyrazolo[3,4-b]pyridines as reactive intermediates in the synthesis of various heterocyclic derivatives by Ashraf A Aly, Talaat I El-Emary, Aboul-Fetouh E Mourad, Zainab Khallaf Alyan, Stefan Bräse and Martin Nieger in Journal of Chemical Research
Footnotes
Acknowledgements
The authors thank the DFG foundation for finanical support for the accommodation of Prof. Ashraf A. Aly during his stay at the Karlsruhe Institute of Technology, Karlsruhe, Germany.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
ORCID iD
Ashraf A Aly
References
1.
YuGMasonHWuX, et al. J Med Chem2001; 44: 1025.
2.
TuckerTJSiskoJTTyneborRM, et al. J Med Chem2008; 51: 6503.
3.
OchiaiHIshidaAOhtaniT, et al. Bioorg Med Chem2004; 12: 4089.
4.
BareTMMcLarenCDCampbellJB, et al. J Med Chem1989; 32: 2561.
5.
XingYZuoJKrogstadP, et al. J Med Chem2018; 61: 1688.
6.
ChuILynchBM.J Med Chem1975; 18: 161.
7.
FeurerALuithleJWirtzS, et al. WO 2004009589, 2004.
8.
GudmundssonKSJohnsBAWangZ, et al. Bioorg Med Chem2005; 13: 5346.
9.
ShutskeGMRoehrJE.J Heterocycl Chem1997; 34: 789.
10.
HenkeBRAquinoCJBirkemoLS, et al. J Med Chem1997; 40: 2706.
11.
StraubABenet-BuckholtzJFrodeR, et al. Bioorg Med Chem2002; 10: 1711.
12.
HuangSLinRYuY, et al. Bioorg Med Chem Lett2007; 17: 1243.
13.
SaggarSASiskoJTTuckerTJ, et al. US Patent 2007021442, 2007.
14.
ZhangPPennellAMKWrightJJK, et al. WO 2007002293, 2007.
15.
ChiuGLiSConnollyPJ, et al. WO 2006130673, 2006.
16.
PatelJBMalickJBSalamaAI, et al. Pharmacol Biochem Behav1985; 23: 675.
17.
BoerrigterGBurnettJC.Cardiovasc Drug Rev2007; 25: 30.
18.
SanghviYSLarsonSBWillisRC, et al. J Med Chem1989; 32: 945.
19.
AlyAAEl-EmaryTIMouradAE, et al. J Heterocycl Chem2019; 56: 1369.
20.
GeraldoRBBelloMLDiasLRS, et al. Thromb2010; 17: 730.
LeuserHPerroneSLironF, et al. Angew Chem Int Ed2005; 44: 4627.
27.
SheldrickGM.Acta Crystallogr2015; A71: 3.
28.
SheldrickGM.Crystallogr2015; C71: 3.
29.
SpekAI.Acta Crystallogr2009; D65: 148.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.