A new and efficient synthesis of 6-bromo-8-cyclopentyl-5-methyl-2-(methylsulfinyl)-pyrido[2,3-d]pyrimidin-7(8H)-one, a key intermediate of Palbociclib, starting from thiouracil was described. This protocol involved methylation, nucleophilic substitution, bromination, nucleophilic substitution, Heck reaction, ring closure, oxidation, and bromination to afford a key intermediate of Palbociclib with approximately 35% overall yield. The advantages of this developed synthetic strategy included improved overall yield, inexpensive starting materials, and readily controllable and cleaner reaction conditions.
Breast cancer (BC) is the most common cancer and leading cause of death in women worldwide. Cellular proliferation, growth, and division are strictly controlled by the cell cycle regulatory machinery. An important pathway is cyclin-dependent kinases (CDKs) which regulate the cell cycle and thus control transcriptional processes.1 Cyclin-dependent kinases 4 and 6 (CDK4/6) are key regulators of the cell cycles that control cellular progression from growth phase (G1) into phases associated with DNA replications (S). Increased CDK4/6 activity is frequently observed in estrogen receptor–positive (ER+) BC.2,3 Palbociclib (Figure 1) is a highly selective, reversible inhibitor of CDK4/6 and could block tumor cell proliferation. Palbociclib was approved by the United States Food and Drug Administration (FDA) in February 2015 and marketed in the name of IBRANCE. It was often employed to treat post-menopausal women having advanced BC with ER+, human epidermal growth factor receptor 2–negative (HER2−) as an initial endocrine-based therapy in combination with Letrozole.4–6
The chemical structure of Palbociclib.
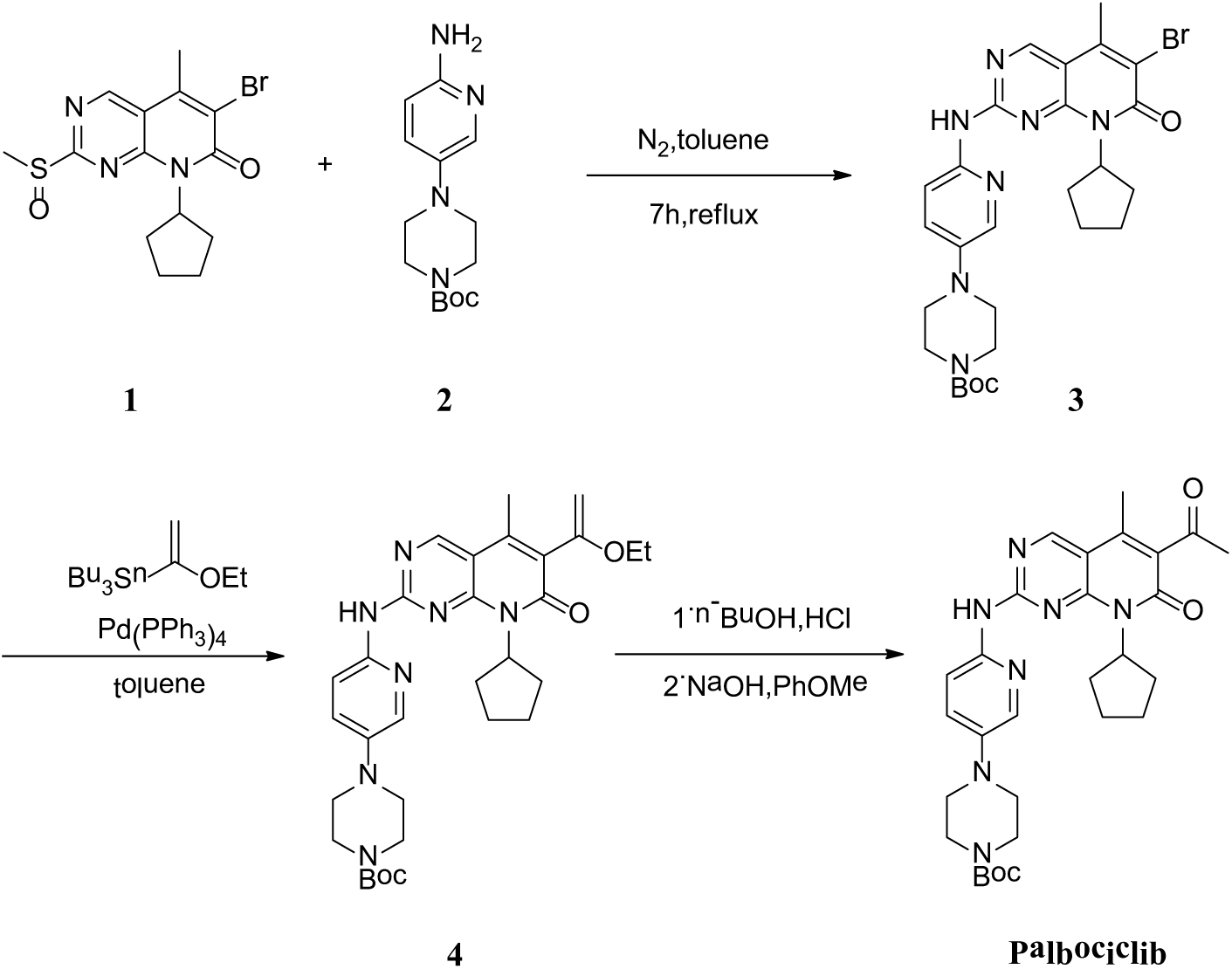
Several methods for the synthesis of Palbociclib have been reported. One of the synthetic strategies mainly involved the nucleophilic substitution reactions of intermediate 1 and intermediate 2, which reacted in toluene at 100 °C to give intermediate 3. Then, intermediate 3 was treated with (1-ethoxyvinyl)tributyltin under Stille conditions to give intermediate 4, which afforded Palbociclib after aqueous workup (Scheme 1).7 Hence, a key to producing Palbociclib is the synthesis of intermediate 1.
The synthetic strategies for the preparation of Palbociclib.
A significant synthetic strategy for the preparation of intermediate 1 was reported in 2005. This protocol started from 4-chloro-2-methylthio-pyrimidine-5-carboxylic acid ethyl ester and involved an eight-step sequence. After displacement of the chlorine of intermediate 5 with cyclopentylamine, a two-step reduction–oxidation sequence converted the ester functional group to an aldehyde, permitting reaction with a Grignard reagent to convert the aldehyde to an alcohol which was oxidized to a ketone with N-methylmorpholine N-oxide (NMO) and tetrapropylammonium perruthenate (TPAP). In the later steps, a Witting–Horner reaction, followed by an N-bromosuccinimide (NBS) bromination reaction, and finally oxidation gave a methyl sulfoxide with 2-benzenesulfonyl-3-phenyl-oxaziradine and yielded intermediate 1 (Scheme 2).7,8 The drawbacks of this method included low yield (approximately 4.6%), expensive reagents, hazardous reagents, and the risk on scale-up of environmental pollution.
The synthetic strategies for the preparation of the key intermediate 1.
Hence, it was necessary to develop an efficient and economical route to synthesize intermediate 1. We have developed a new synthetic strategy for intermediate 1 (Scheme 3). Methylation of thiouracil, nucleophilic substitution with SOCl2, bromination, nucleophilic substitution with cyclopentylamine, a one-pot two-step method (Heck reaction, ring close sequence), and oxidation of the methylthio group at the 2-position while introducing bromine gave intermediate 1. This strategy used dimethyl carbonate to replace dimethyl sulfate and methyl iodide as the methylation reagent in the first step to avoid environmental pollution and used thionyl chloride to replace phosphorus oxychloride as the reagent in the second step to reduce the difficulties of post-processing. This strategy used a Heck reaction in a ring-closing sequence to shorten the number of reaction steps and improved access to intermediate 11 via the fifth step. This route usefully oxidized the 2-position methylthio group to a sulfoxide while introducing the bromine with NBS in the sixth step and thus shortened the number of reaction steps and avoided the use of expensive reagents.
The new synthetic strategies for the preparation of the key intermediate 1.
Results and discussion
The determining step in the route was the procedure for synthesizing intermediate 1, obtained in approximately 34.7% overall yield from thiouracil permitting the valuable synthesis of intermediate 1 starting from commercial thiouracil (Scheme 3).
The route began with the methylation reaction between thiouracil and dimethyl carbonate being tried first. Methods for thioether methylation in industry and academia often employed hazardous and toxic reagents, such as iodomethane and dimethyl sulfate.9–11 Therefore, we wished to use dimethyl carbonate to replace toxic dimethyl sulfate and methyl iodide as the methylation reagent in the first step.12,13 In the presence of potassium carbonate and a phase transfer catalyst, we adopted the method of batch loading in which dimethyl carbonate was slowly added dropwise when the reaction temperature was raised to 120 °C. This method afforded intermediate 14 in 92.6% yield.14–18 A further advantage of this procedure was the environmental benefit.
In the second step, we used thionyl chloride to replace phosphorus oxychloride.19 This method reduced the difficulty of post-processing. In the third step, bromination of intermediate 15 in a mixed solution of MeOH/MeCN (1.4:1) gave intermediate 16 with NBS.
In the fourth step, initially, we hoped that Et3N could catalyze this reaction (Table 1, entry 1). However, it failed to produce intermediate 17.20–22 Then, we screened several other basic catalysts including MeONa, EtONa, and N,N-diisopropylethylamine (DIPEA). It was found that only a small amount of product was obtained at room temperature using DIPEA as the base (Table 1, entries 1–6). Subsequently, by raising the reaction temperature from 25 to 100 °C, it was found that DIPEA as the base successfully improved the yield from 46.5% to 90.4% (Table 1, entries 7–10).
NR: no reaction; NP: no product; DMF: dimethylformamide; THF: tetrahydrofuran; DIPEA: N,N-diisopropylethylamine.
4-Chloro-5-bromo-2-(methylthio)pyrimidine (10 mmol) and cyclopentylamine (15 mmol).
Isolated yield.
The fifth step, known as the Mizoroki–Heck reaction, is a coupling reaction of an unsaturated halogenated hydrocarbon with an olefin to form a substituted olefin using a strong base with palladium catalysis and nitrogen protection. In a previous report, intermediate 11 was obtained by the Witting–Horner reaction with (EtO)2P(O)CHXCO2Et (Scheme 2).7,8 This strategy used a Heck reaction and amidation reaction to give intermediate 11 (Scheme 3). Typical palladium (II) catalyst compounds included Pd(OAc)2, Pd(PhCN)2Cl2, PdCl2, and Pd(dppf)2Cl2. Typical phosphine ligand compounds used for the Heck reaction included monodentate phosphine agents such as PPh3, (o-CH3Ph)3P, and 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP). Typical bases mainly included triethylamine (TEA), DIPEA, K2CO3, and sodium acetate.23–25 Palladium catalysts, ligands, and bases had interesting effects on the reaction. Initially, we used Pd(OAc)2 and PPh3 which might catalyze this reaction using Et3N as the base. However, no reaction was observed (Table 2, entry 1). After screening the catalyst system, we successfully used Pd(PhCN)2Cl2 as a palladium catalyst, DIPEA as a base, and (o-CH3Ph)3P as a carrier to give intermediate 11 with crotonic acid under nitrogen protection (Table 2, entries 2–6). We also examined this catalyst system at different temperatures to improve the yield (Table 2, entries 6–8). Then, we successfully improved the yield to 76.3% at 130 °C.
The sixth and last steps involved oxidation at the 2-position of the methylthio group while introducing bromine. It has been previously shown that aromatic sulfides are oxidized cleanly to sulfoxides in aqueous media when treated with NBS.26,27 Tagaki et al.27 have demonstrated that certain aromatic sulfides could be oxidized to sulfoxides in 70% dioxane-containing water containing NBS in 1964. In addition, Surendra et al.’s28 research also demonstrated the oxidation of sulfides to sulfoxides by β-cyclodextrin in water under neutral conditions in 2005. Based on the previous literature and Surendra et al.’s studies, we designed a reaction system of intermediate 11/NBS/water/AcOH (1:3:1:0.1), which could oxidize the 2-position methylthio group while introducing bromine without any over-oxidation to sulfones. This method not only avoided long reaction times and hazardous reagents, but also shortened the reaction steps and increased the yield of intermediate 1. In conclusion, a simple and efficient six-step method for synthesizing an important intermediate 1 of Palbociclib from commercially available thiouracil is described. In contrast to the procedure reported in the literature, this method offered a route based on good yields, with low cost, readily controllable reaction conditions, and reduced environmental hazards. This procedure has potential for scale-up in production.
Experiment
The solvents, reagents, and materials were commercially available and were used without further purification unless otherwise noted. All reactions were monitored by thin-layer chromatography (TLC) on silica gel plates (GF-254; Qindao Ocean Chemical Company, China) and silica gel column chromatography (CC) (200–300 mesh; Qingdao Marine Chemical Industry Corporation, China). Melting points (m.p.) were determined on a YRT-3 drug melting point meter and were uncorrected. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance DPX-300 MHz/500 MHz instrument in CDCl3 with tetramethylsilane (TMS) as an internal reference and the chemical shifts (δ) were reported in parts per million (ppm). High-resolution mass spectra (HRMS) were obtained from Agilent 1100 LC/MS Spectrometry Services.
2-Methylthio-4-ketopyrimidine (14)
To a solution of thiouracil (13, 10 g, 78.1 mmol), tetrabutylammonium bromide (12.59 g, 39.1 mmol), and K2CO3 (16.2 g, 117 mmol) in dimethylformamide (DMF), dimethyl carbonate (30 mL) was slowly added dropwise when the temperature was raised to 120 °C. After addition, the mixture was stirred at 120 °C for 6 h. After filtering K2CO3, water was added to the filtrate which was then extracted with ethyl acetate. The organic phases were combined, dried over Na2SO4, and concentrated in vacuo. The residue was purified by passing the organic extract through a silica gel column using dichloromethane (DCM)/MeOH (75:1) to give the product 14 as a white solid (10.27 g, 92.6%); m.p. 204.4–205.3 °C. 1H NMR (300 MHz, CDCl3) δ 7.88 (d, J = 6.5 Hz, 1H), 6.23 (d, J = 6.6 Hz, 1H), 2.58 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 164.7, 162.8, 155.0, 111.1, 13.4; HRMS ([M + H]+): m/z calcd for C5H6N2OS: 143.0279; found: 143.02739.
4-Chloro-2-(methylthio)pyrimidine (15)
To a solution of 2-methylthio-4-ketopyrimidine (14, 10.27 g, 70.4 mmol) in DCM (20 mL) and DMF (10 mL) mixed solution, thionyl chloride (3.05 mL, 42.2 mmol) solution was slowly added dropwise while heating. After addition, the reaction mixture was stirred at 40 °C for 3 h. After quenching with saturated NaHCO3, the solution was adjusted to a neutral pH and the solution was extracted three times with DCM. The organic phases were washed with saturated NaHCO3 solution and saturated NaCl solution and dried over anhydrous Na2SO4. After filtering to remove the anhydrous Na2SO4, the solution was distilled at 40 °C to remove solvents under atmospheric pressure to give product 15 as a yellow oil (10.19 g, 87.6%). 1H NMR (300 MHz, CDCl3) δ 8.38 (d, J = 5.2 Hz, 1H), 6.99 (d, J = 5.2 Hz, 1H), 2.53 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 174.0, 161.0, 158.0, 116.4, 14.3; HRMS ([M + H]+): m/z calcd for C5H5ClN2S: 160.9940; found: 160.99335.
4-Chloro-5-bromo-2-(methylthio)pyrimidine (16)
To a solution of 4-chloro-2-(methylthio)-pyrimidine (15, 10.19 g, 63.4 mmol) in MeOH (28 mL) and MeCN (20 mL) mixed solution, NBS (13.55 g, 76.1 mmol) was added (2 × 3.76 g). After the additions, the reaction mixture was stirred at room temperature. After quenching with saturated Na2SO3 solution, the solution was extracted with DCM and saturated NaHCO3 solution three times. The combined organic phases were dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by passing the organic extract through a silica gel column using PE/EA (25:1) to give the product 16 as a white solid (11.02 g, 72.5%); m.p. 48.5–50.1 °C. 1H NMR (300 MHz, CDCl3) δ 8.54 (s, 1H), 2.55 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 172.1, 159.5, 114.1, 14.7. HRMS ([M + H]+): m/z calcd for C5H4BrClN2S: 240.9025; found: 240.90203.
To a solution of 4-chloro-5-bromo-2-(methylthio)pyrimidine (16, 11.02 g, 46.0 mmol) in dioxane (40 mL), DIPEA (8.15 mL, 46.0 mmol) and cyclopentylamine (5.44 mL, 55.2 mmol) were added into the solution. After completion of the addition, the reaction mixture was stirred at 100 °C for 4 h. After quenching with water, the solution was extracted with ethyl acetate and saturated NaHCO3 solution three times. The combined organic phases were dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by passing the organic extract through a silica gel column using PE/EA (15:1) to give product 17 as a yellow oil (11.99 g, 90.4%). 1H NMR (300 MHz, CDCl3) δ 8.03 (s, 1H), 5.24 (s, 1H), 4.38 (dd, J = 13.6, 6.8 Hz, 1H), 2.48 (s, 3H), 2.09 (td, J = 11.7, 6.0 Hz, 2H), 1.77–1.62 (m, 4H), 1.49 (dt, J = 12.3, 6.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 170.3, 157.1, 154.8, 99.8, 52.9, 23.8, 14.4; HRMS ([M + Na]+): m/z calcd for C10H14BrN3SNa+: 311.9969; found: 311.99651.
To a solution of 5-bromo-N-cyclopentyl-2-(methylthio)pyrimidin-4-amine (17, 11.99 g, 41.6 mmol) and crotonic acid (5.37 g, 62.4 mmol) in DMF (100 mL), DIPEA (29.46 mL, 166.4 mmol) was added. Then palladium dibenzylcarbonitrile (478 mg, 1.2 mmol) and tri-o-tolylphosphorus (886 mg, 3.0 mmol) were added under a nitrogen atmosphere. After completion of the addition, the reaction mixture was stirred at 130 °C for 24 h under nitrogen protection. After 24 h, acetic anhydride (15 mL) was added and the solution was stirred at 70 °C for 4 h. Then the mixture was cooled, diluted with methyl tert-butyl ether (MTBE; 100 mL) and then extracted with NH4Cl, NaHCO3, and water. The organic phases were combined, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by passing the organic extract through a silica gel column using PE/EA (15:1) to give product 11 as a gray solid (4.06 g, 76.3%); m.p. 201.2–202.4 °C. 1H NMR (300 MHz, CDCl3) δ 8.68 (s, 1H), 6.41 (s, 1H), 5.97–5.82 (m, 1H), 2.61 (s, 3H), 2.36 (d, J = 20.9 Hz, 5H), 2.06 (s, 2H), 1.87 (s, 2H), 1.69 (d, J = 10.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 163.0, 154.1, 144.0, 122.0, 111.0, 53.8, 28.4, 25.8, 17.2, 14.5.; HRMS ([M + Na]+): m/z calcd for (C14H17N3OSNa+): 298.0990; found: 298.09853.
To a solution of 8-cyclopentyl-5-methyl-2-(methylthio)pyrido[2,3-d]pyrimidin-7(8H)-one (11, 4.06 g, 14.7 mmol) in MeCN (30 mL), acetic acid (84 µL, 1.47 mmol), H2O (265 µL,14.7 mmol), and NBS (7.87 g, 44.2 mmol) were added. After completion of the addition, the reaction mixture was stirred at room temperature for 8 h. After quenching with water (70 mL), the solution was stirred for 10 min and filtered. The filter cake was washed with water to obtain a white solid. After purification by recrystallization with ethanol (50 mL), the product was dried in vacuo to give product 1 as a white solid (4.69 g, 85.6%); m.p. 158.3–160.4 °C. 1H NMR (300 MHz, CDCl3) δ 8.79 (s, 1H), 6.04 (t, J=8.8Hz, 1H), 3.48 (s, 3H), 2.62 (s, 3H), 2.28(dt, J=14.7, 7.3Hz, 1H), 2.10 (t, J= 7.7Hz, 1H), 1.90(dd, J = 13.2, 7.7 Hz, 1H)1.72-1.61 (m, 1H),1.42(s, 1H), 1.29(t, J=11.6Hz, 4H); HRMS ([M + Na]+): m/z calcd for (C14H16BrN3O2SNa+): 394.0024; found: 394.00248.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was partially supported by the Fundamental Research Funds for the Central Universities (No. 2242014R30019).
References
1.
BilginBSendurMAŞenerDDet al. Curr Med Res Opin2017; 33: 1559.
2.
GuptaAKSharmaSDahiyaNet al. Med J Armed Forces India2016; 72: S37.