
Abstract
Although the use of the cluster randomized trial (CRT) design to evaluate vaccines, public health interventions or health systems is increasing, the ethical issues posed by the design are not adequately addressed, especially in low- and middle-income country settings (LMICs). To help reveal ethical challenges, qualitative interviews were conducted with key stakeholders experienced in designing and conducting two selected CRTs in Malawi. The 18 interviewed stakeholders included investigators, clinicians, nurses, data management personnel and community workers who were invited to share their experiences related to implementation of CRTs. Data analysis revealed five major themes with ethical implications: (1) The moral obligation for health care providers to participate in health research and its compensation; (2) Suboptimal care services compromising the integrity of CRT; (3) Ensuring scientific validity and withholding care service; (4) Obtaining valid consent and permission for waiver of consent; and (5) Inadequate risk assessment for trial participation. Understanding key ethical issues posed by CRTs in Malawi could improve ethical review and research oversight of this particular study design.
Background
Although cluster randomized trials (CRTs) are increasingly used to evaluate vaccines, public health interventions and health systems, the best practices for ethics review and research oversight are nascent and underdeveloped, especially in low- and middle-income counties (LMICs) (GFBR, 2017; Hunt et al., 2019; Joag et al., 2019; Mtande et al., 2019; Osrin et al., 2009; Taljaard et al., 2011; Weijer et al., 2011). The developers of the Ottawa Statement on the Ethical Design and Conduct of CRTs clearly indicated that ethical considerations arising in LMIC settings are not comprehensively addressed (Weijer et al., 2012). The authors of the Statement note, “LMIC perspectives were under-represented (and). . .we recommend that subsequent revisions include greater LMIC representation.”
While the Council for International Organizations of Medical Sciences (CIOMS) Guideline 21 is devoted to the ethical conduct of CRTs, (CIOMS, 2016), the guidance is largely drawn from the Ottawa Statement. Shortcomings in guidance are important because they might expose participants to undue risks, while on the other hand, strict enforcement of conventional rules to CRTs can pose unnecessary obstacles, undermine scientific quality or impede improvements in patient health and health system outcomes (Taljaard et al., 2018). CRTs are not alone in this regard; the ethical implications of many alternative clinical trial designs, including CRTs in LMICs have not received sufficient attention from bioethicists, researchers, and policymakers (Hunt et al., 2019).
The 2017 Global Forum on Bioethics in Research (GFBR) and others (GFBR, 2017; Joag et al., 2019; van der Graaf and Cheah, 2019) did highlight some ethical issues posed by CRTs conducted in LMICs settings. From the GFBR (2017) conference we learn that further guidance is required for (1) concerns related to cluster selection; (2) valid consent and consent waiver; (3) incentives and coercion; (4) community acceptability; (5) obligations of researchers, sponsors, and government for post-trial access; and (6) health record linkage and big data use for health research. But most CRT researchers report trial results only, while key ethical issues posed by the trial design are inadequately reported (Taljaard et al., 2011).
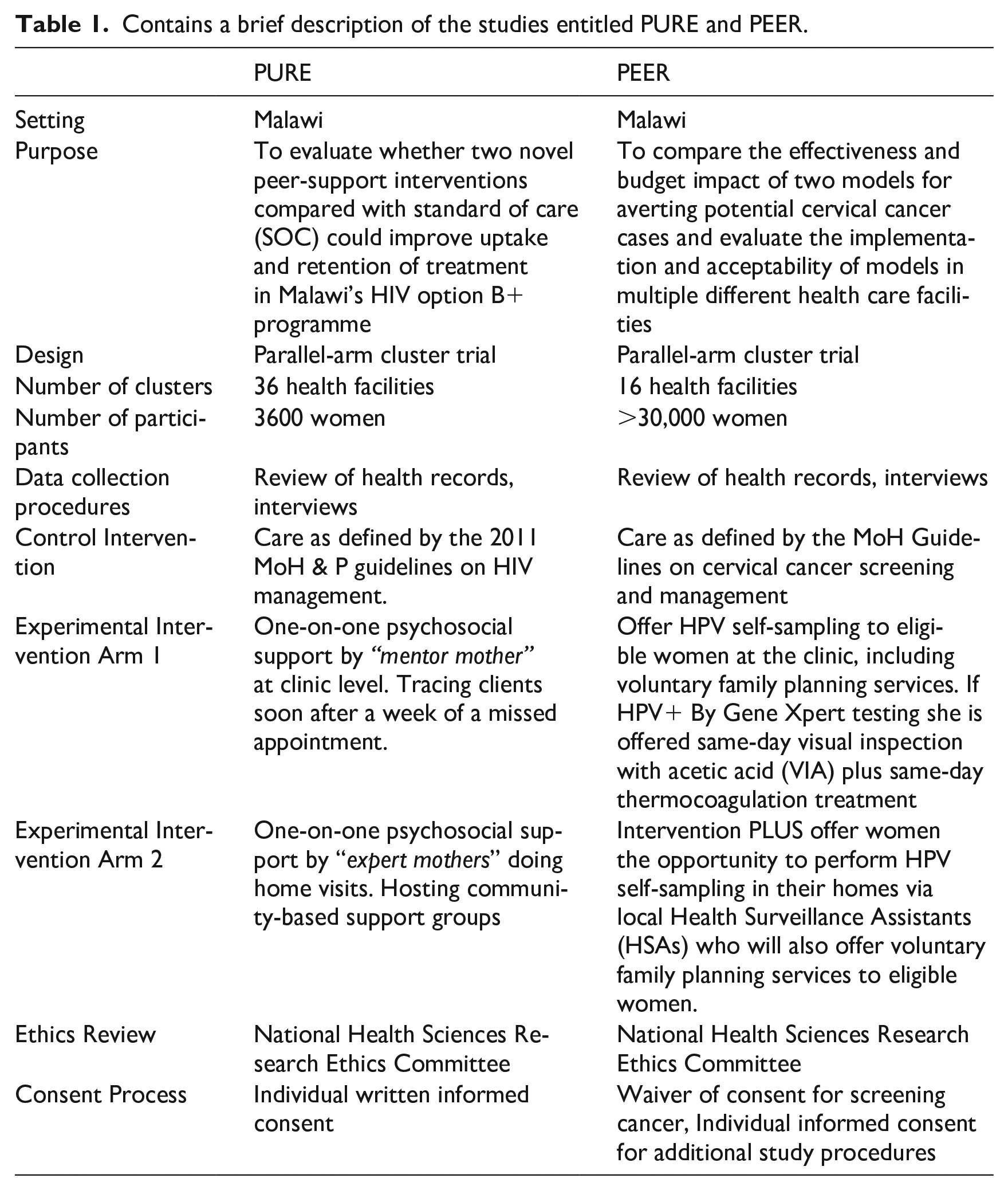
The identification of key ethical issues, grounded in the experiences of those directly involved in the design, implementation, and evaluation of the trials is a promising way to develop appropriate guidance for CRTs (Nicholls et al., 2018). With this in mind, a multiple case study design with qualitative methods was used to explore key ethical issues posed by selected cases conducted in Malawi’s public health care setting. In-depth interviews were conducted with key stakeholders involved in the implementation of two CRTs. The two studies varied in terms of consenting process. Table 1 Contains a brief description of the studies entitled PURE and PEER.
Contains a brief description of the studies entitled PURE and PEER.
Methods
Data collection
The study was reviewed and approved by two research ethics committees (RECs): the National Committee on Research in Social Sciences and Humanities in Malawi and the Health Research Ethics Committee at Stellenbosch University in South Africa. The Principal Investigators of the selected cases provided permission to interview team members and individual informed consent was obtained. Interviews were conducted in person or via telephone by one researcher (TKM) with training and experience in qualitative methods and research ethics. Interview guides were adapted from a study by Nicholls and colleagues (Nicholls et al., 2018). In the present study, the Nicholls et al. (2018) interview guides were modified to focus on ethical issues posed by pragmatic CRTs (see Supplemental Materials). The full guide comprised of three main sections: (1) role in the selected trial; (2) experiences of ethical issues arising during the design and conduct of the trial and (3) perspectives on appropriate oversight and regulation of CRTs. Key informants from the PURE team (N = 10) and from the PEER team (N = 8) were interviewed between September 2021 and August 2022. The interviewees were purposively selected, and snowball sampling was also used. Interviews were audio-recorded and transcribed verbatim by an experienced research assistant. Transcripts were checked for accuracy and consistency. Final versions of transcripts were imported into qualitative data analysis software (Atlas ti. 22) for analysis.
Analytic methods
Using a content analysis approach (Hsieh and Shannon, 2005), codes were developed after reading the transcripts from each case. We drew up an initial list of codes using a hierarchical tree structure with three levels of codes, based on pragmatic trial domains from the interview guide. All transcripts were grouped into two categories (PURE and PEER). Searches were run on each code and reports were generated. Thematic analysis (Braun and Clarke, 2006) was used to manually categorize the data into a finer set of themes and findings. Interim and final analyses were presented to the broader research team for discussion. A theme was denoted an ethical theme if it was related to the safety, welfare and rights of participants and/or the research community, and if it was related to CRT design.
Findings and discussion
Eighteen interviews were conducted with investigators, clinicians, data management personnel, nurses and community workers. The interviewees were comprised of staff working in organizations that designed the CRTs, staff that provided technical support and staff from public health facilities that delivered study procedures. Discussions drew on a breadth of experiences and roles in the trial. Some participants were involved during protocol development while others joined the trial mid-way. An overview of participant characteristics is presented in Table 2.
Interviewed stakeholders.

After analysis of the interview data, we identified five ethically relevant themes from the respondents’ experiences and opinions about the ethical challenges posed by CRTs in public health facilities. Our findings are consistent with what has been reported in literature (Chaudhry et al., 2013; GFBR, 2017; Goldstein et al., 2018; McRae et al., 2013; Nicholls et al., 2020). However, these ethical challenges manifest themselves differently in the unique environment of health facilities in Malawi. The purpose of this qualitative study was to describe key ethical issues; how best to resolve the ethical challenges that were identified is beyond the scope of this article. In what follows, we discuss each theme in turn, and provide illustrative quotes for each. Considering that the CRTs are identifiable, direct quotations from respondents have not been tagged by profession, role or study name to maintain confidentiality. The quotations themselves have not been changed.
Ethical themes
The moral obligation for health care providers to participate in health research and its compensation
This theme raises the question of whether clinicians have the moral duty to participate in pragmatic clinical trials (Garland et al., 2023). Garland and colleagues have skillfully mapped the moral obligation to participate in such trials as well as certain exceptions. According to Garland et al., expecting (but not receiving) personal benefits is not considered a valid exception for refusing participation in a pragmatic trial. In contrast, our data suggests that the issue of “personal benefit” needs careful deliberation to promote the study design in this setting.
Interviewees reported that, for some facilities, the health providers felt that the potential benefits of participation were inadequate, so they unanimously agreed not to be included as a trial site. And in other sites, the health providers accepted and went through protocol training, yet they never implemented the study procedures. Despite engaging the Ministry of Health leadership at different levels, health providers in the public facilities were at liberty to participate or not participate and their decisions were respected.
“Some providers chose not to participate, and we honored their choices”
Stillmore there were efforts to improve acceptability to participate in research. The investigators reported that introducing the research as a “project” or “program” to the health providers promoted ownership and increased acceptability.
“We were working with health care workers, so when you say this is a study, they think about someone should get paid to do study procedures, so we were trying to run away from that mentality because we did not want to give extra pay.[. . .] But using the word ‘project’ we felt it was something that they could own and it was just a better wording for us but it was definitely a research study. . .”
But this raises the question of whether it is ethically right to frame a CRT in this way, especially if it risks confusing patients about study participation. Moreover, if the CRT involves implementation science, are the HCWs then research participants? How best do we compensate HCWs to promote and implement these trials? Accurate identification of research participants, compensation in research, and voluntary participation are important aspects of the responsible conduct of research. The findings of our study revealed that fair distribution of burdens and benefits between HCWs and investigators in a CRT needs further examination.
Most interviewees in both studies also commented that participating in a CRT means additional work for which they should be compensated. Although the incentives acted as a motivation to many health providers, the concern was that the mode of payment caused strife too. In one study, not all providers at the facility hosting the trial were receiving the incentives. The health providers who were not on the list for incentives were not cooperative with respect to patient care and study implementation:
“. . .. So, of course people had issues but not much, the issues were that someone would say “I will not do some of the procedures. Send this participant[patient] to that staff, that is the person involved with that. They would refer the person to someone who is receiving the incentives and the procedure would still be done”
By contrast, some interviewees commented that they were not given incentives as an individual, but it was given for the entire health facility. This mode of payment was arrived after a careful deliberation within the study team. They concluded that all health providers present at a facility were contributing to the study, to some extent. Therefore, the leadership of the randomized facility was asked to decide the items to buy to benefit them all:
“. . .The members providing the technical support informed us that they have a budget allocation to give incentives, but it is up to us to decide how to use the money. They were supporting us with gloves and sample collection materials, but they deliberately arranged this special incentive. I think they wanted to motivate us. . .”
Suboptimal care services compromising the integrity of CRT
The theme in this section is about care services which can compromise CRT integrity, especially when care delivery is suboptimal. Most public health facilities in Malawi are understaffed, have infrastructure challenges and are ill-equipped such that the quality of health care delivery is often affected (Makwero, 2018) as well as adherence to ethical principles. The local de facto standard of care may be significantly different than de jure standard of care (London, 2002). CIOMS (2016) Guidelines provides that participants in the control group of a trial must receive an established effective intervention. In cases of suboptimal care services, should low performing sites therefore be excluded from participation in a CRT? How might this affect generalizability of the study findings? Other investigators may include a substantial budget for improving care services in public facilities, but this raises a question as to how much improvement can be made before undermining the validity of the CRT data.
Most Interviewees from both studies reported that that what the policy or guidelines stipulate is often different from the actual care service delivered in routine settings and that the standards of care vary across clusters. Some interviewees from one study wondered whether improving the services would adversely affect the relevance of the study, and also whether excluding low performing sites was ethical. Furthermore, in terms of privacy, interviewees noted that if strict privacy rules in research were to apply, it would not have been possible to conduct their trial. They commented that practices that undermine patient privacy and confidentiality were part of usual care, and they were adopted when conducting research.
“I went to a facility, and it is a big facility but HIV positive people are being registered on a veranda and they are getting their details there, so if you do a study, you will not change the location, you will register them at the same place because there is no room allocated, so it’s either you do that or you don’t do the study there [. . . . .] I don’t know what one can do about that. You are on the site to do the study, but you are also concerned that there are these ethical issues and if I do the study, I will be considered to have breached ethical consideration, but if you get into the details, that is the practice there! Is it not? Will it not be?”
By contrast, some interviewees commented that their study budget included funds for renovations in all trial sites, including the connection of electricity, and the renovations included demarcation of rooms and procurement of curtains to improve privacy. Concerns were raised regarding a substantial study budget allocated to health care improvements; one interviewee commented that Malawian RECs may need to consider a waiver of the usual 10% levy that is collected from CRT research projects, since it is difficult to conduct CRTs without making some improvements in public health facilities. Similarly, most interviewees raised concerns related to the data management infrastructure, staff shortages and high staff turnover in public facilities which affects the delivery of study procedures. More specifically, some interviewees mentioned that some health providers who are new to research practices tend to make many mistakes, including enrolling ineligible participants.
“let’s say we train a clinician today, within no time you find that they have transferred to another clinic. We had invested a lot in terms of training, for them to understand the protocol then they go. The new staff would come, they begin to do their own things, more less like guess work. So, that it would contribute to recruiting participants which are not eligible.”
Ensuring scientific validity and withholding care service
The third theme is related to the second theme above, but different because it focuses on the question of harm: is it right to withhold care services from current patients because there is a very important trial being conducted that could benefit future patients? In the Malawian setting, there are many partner organizations who implement quality improvement programs in public health facilities. Their services may not be considered established effective interventions per CIOMs guidelines (CIOMS, 2016), yet they have the potential to improve health care delivery. The overarching question is: how can scientific rigor in a CRT be maintained while avoiding harms due to withholding such improvement programs from CRT sites? The concern expressed in our findings confirms what was reported by Rosenberg et al. (2014) on how CRT researchers struggle to ensure scientific rigor: “. . .one standard of care site was found to have a community-based program that was missed during the situational analysis. Another site is planning new interventions, despite requests to delay until study completion. During the follow-up period, additional sites may make similar decisions, potentially leading to bias.” (p. 8)
Some interviewees reported that the investigators wanted to make sure they were obtaining valid trial results, without any interference. However, other partner organizations were implementing health services that could potentially improve the standard of care arm. In liaison with the district health management team (DHMT) they transferred out these organizations. Some of them relocated to other health facilities which were not participating in the trial, while others were asked to implement different health service from what was initially proposed.
“the DHMT would suspend those and allocate them to another site and we continued [. . .] but we made sure that those programmes should not continue in those study sites. We took note of the time they started because we wanted to adjust our results based on the dates those interventions were started. The trial continued because we had already invested in those sites. We continued but they stopped. The moment we had an interface meeting with DHMT they stopped[. . .] And it was not only one programme, we had others like [names withheld]”
By contrast, some interviewees reported that, upon realizing there were other health partners with similar interests, they decided to collaborate. But there were others with whom it was difficult to collaborate. In such cases, they delayed study procedures for that specific trial site to let the other health partner organization finish their project.
These findings show that researchers delayed the implementation of study interventions in some trial site(s) to allow another partner organization to finish their quality improvement services. In terms of a parallel CRT design, is this delay acceptable? The use of reliable scientific methods is a basic ethical requirement of health research. Unreliable methods risk producing invalid data, wasting research resources, and potentially harming individuals and communities if the data are to inform health policy. Clear guidance on this point is relevant to low resource settings with significant variations in standards of care between clinical sites.
Obtaining valid consent and permission for waiver of consent
Despite the key considerations provided in the Ottawa statement and the CIOMS (2016) Guidelines for waiving consent in CRTs, our findings indicate that the Malawian RECs are strict around permission for waiver of consent. Concerns indicate that health providers were overstretched and, due to time constraints, sometimes conducted group consent. Considering the routine care service delivery, if health providers conduct consent processes for CRTs, is this not contributing to differential care between research participants and ordinary patients? (Morain et al., 2019). According to our findings, the ethical requirements for informed consent are being missed, but the legal aspect of consent is probably being satisfied. Moving forward, the question arises as to how best should RECs review a recruitment plan for CRTs, including the consent process?
One interviewee who was involved with the initial ethics application to the REC commented that the researchers requested a waiver of consent from their Malawian REC. However, the researchers’ request was rejected, and they were instructed to develop an individual consent form to be administered in the trial. The interviewees who were involved with enrollment of participants commented on the practical difficulties (namely staff shortages and high workload) for obtaining valid individual consent, to the extent that health providers conducted group consent sessions:
“. . .maybe there would be four clients who you would want to enrol with each one of them requiring privacy and to be consented individually, the concern is that we were not doing it in the way that it is supposed to be done, privacy in terms of both visual and audio. [. . .] We were supposed to sit in a separate room so that we explain to the person in a way that no one else hears what is happening.”
Some interviewees who were enrolling participants commented that due to power imbalance between the health providers and the patients, most of the patients agreed to enroll in the trial. The interviewees reported that they were trained to observe the International Conference on Harmonization - Good Clinical Practices (ICH-GCP) guidelines, but it was not possible to follow the recommendations. They felt that the level of interventions facilitated the thinking that enrollment in the trial was mandatory, and some health providers had difficulties in separating research from routine practice. Consequently, some patients did not receive the routine care service because they had refused trial participation:
“we could find that the register has recorded a case, but was not initiated on treatment, and then you ask them they say ‘oh they refused to enroll in the study!’ And you are like ‘hmm the two are different, they could have been initiated the treatment even if they don’t want to participate in the study.’ So, the main challenge was to differentiate the two. We embedded the study inside clinical care”
They also commented that study participants who were recruited to a sub-study which involved in-depth interviews came to learn that they were trial participants later. A minority of participants subsequently withdrew from trial participation, while other participants were well-informed about the CRT and adherence to study procedures improved: “I should mention here that they were few, they were not many! but then we explained the whole process to them and told them that they had the choice to continue or withdraw from the study. So, we asked them if they would like to continue in the study. I think we had very few people who withdrew, it can’t be up to five who had withdrawn after that. . .. But now most of them were participating the study with full information on what is happening. . .”
Some interviewees from one study commented that they sought a waiver of consent from the REC and then were obtaining individual written consent for specific study procedures. They reported that researchers debated extensively to arrive at the decision to seek a waiver of consent since permission from RECs for consent waivers is challenging. In addition, one interviewee commented that the ethics approval letter included a statement that the REC strongly felt that the CRT was a “project” and not a research study and therefore did not require consent procedures.
Our findings show that the REC informed the researchers that that the CRT protocol is not research but a program or project. Could this be a reason that implied consent was adopted in a CRT involving a new vaccine in Malawi? (Doshi, 2020). These findings suggest there is a need to raise awareness regarding the conditions that can warrant a waiver of consent in CRTs. On the other hand, these findings confirm that in pragmatic clinical trials, RECs and researchers often struggle to distinguish research, quality improvement programs and clinical practice, that is, what does and does not require research ethics oversight (Goldstein et al., 2018; Taljaard et al., 2018; Weinfurt et al., 2017).
Inadequate risk assessment for trial participation
The fifth theme concerns inadequate risk assessment in CRTs. Interviewees from both studies were sharing what others have also identified, that the risk in CRTs is often associated with the nature of the intervention (Taljaard et al., 2018). The CIOMS (2016) guidelines provides that aggregate risks of all research interventions or procedures in a study must be considered appropriate in light of the potential individual benefits to participants and the scientific social value of the research. However, our findings show that it was difficult to assess whether the potential harms are “minimal risks” or “more than minimal risks,” since interventions are already part of routine care.
With respect to identifying relevant risks of participating in the trial, the interviewees from both studies noted that the risks may not be obvious during protocol development. One interviewee commented that in their CRT they were evaluating two health system interventions that were already in routine care settings. To their surprise, harms related to a breach of privacy and confidentiality were prominent than what is reported during routine care:
“But the other thing I think is to look into these interventions in detail, like what are they really composed of and how does it affect participants confidentiality or freedom. . . you can have an intervention but if you just read the paper and don’t think outside the box you would say “ah this is okay, because they already do this as a programme”, but I think there are some things in the programme which are not good, so how do we address them when we are conducting the study? Because if the things are done in the routine programme no one gets worried, but if they are done in research, it’s a big issue and I think to maintain the standards of research, this has to be looked into critically.”
Furthermore, most interviewees reported that they thought the risks associated with CRT participation were very much related to the nature of the disease under investigation and the intervention. For example, positive HIV status can expose people to extreme risks from stigma and discrimination:
“. . . . And now everyone in the community knew that these women were following up a particular cohort of people then people were shunning away from their services. They knew that if they begin to get associated with them then it means people will know they have HIV. And in some situations, we had families which were breaking up because they visited them. You know that element of stigma and discrimination. Yes, they were doing home visits so when they go to a house where the mother had not yet disclosed the HIV status to the spouse, . . . so that triggered reaction from spouses like ‘so, what’s happening here?’ We had such scenarios where some marriages even broke-up.”
Some interviewees commented that there are added risks if vulnerable populations are recruited due to the broad inclusion criteria of pragmatic clinical trials. Specific references were made to risks associated with enrolling illiterate populations who were coming from far places to receive services at the public health facilities. The illiterate participants were signing for consent with no appropriate witness, and the intervention providers did not have the capacity to monitor them in case of social harms:
“. . .by broadening the eligibility criteria, we were already raising more ethical issues for instance aah, like the only exclusion criteria was the age, so there was no consideration of the geographical location. . . .And, for people which are not literate, for example consenting these requires a witness, but the witness was someone at the same health center. You can see that it is unethical, but now with the limited number of staff in our public facility we were having illiterate participants sign, sometimes the consenting is done without a witness or a witness is someone who may be biased towards ensuring that the study recruits more”
Most Interviewees from both studies confirmed the presence of social harms and that there were no external study monitors like those in individually randomized drug trials. Neither was there a data and safety monitoring board (DSMB). Some interviewees commented that while all studies had advisory committees, these committees were not functioning as a DSMB.
These issues raise questions about how to best improve the safety of participants involved in CRTs because vulnerable groups are likely to be enrolled. Ordinarily, individual randomized clinical trials involving pharmaceutical companies would hire external monitors, but the trend is different for CRTs.
Final thoughts
These findings must be considered within the limitations of the study. First, the interviewer conducted in depth interviews while working with one of the Malawian RECs. Consequently, respondents may have been uncomfortable discussing ethical issues, especially if they perceived this study as a fault-finding mission. To try to mitigate this impact, the interview guide was shared in advance and extensive efforts were made to establish rapport and build trust before scheduling an interview. Second, purposive selection, coupled with snowball sampling, were used in this study and this may have introduced an element of selection bias. Nevertheless, we believe that interviewees expressed their diverse experiences with CRTs faithfully and the ethical issues that were identified are prevalent in the Malawian context. Our next project includes a normative analysis of the ethical issues raised in this paper to support the development of an ethics assessment tool for CRTs conducted in Malawi.
CRT design is very useful when evaluating an intervention, and this study design is particularly relevant for resource-constrained settings with fragile health services infrastructure in need of improvement. Strengthening ethics evaluation of CRT protocols will promote rights, safety, and welfare of research participants. Characterizing key ethical issues is a promising way to improve ethics evaluation and research oversight of CRTs. Moreover, improving the ethical conduct of CRTs could enhance trust among patient communities and health care providers, which is essential for the successful implementation of these pragmatic trials.
Supplemental Material
sj-docx-1-rea-10.1177_17470161231191247 – Supplemental material for Stakeholders’ experiences of ethical challenges in cluster randomized trials in a limited resource setting: a qualitative analysis
Supplemental material, sj-docx-1-rea-10.1177_17470161231191247 for Stakeholders’ experiences of ethical challenges in cluster randomized trials in a limited resource setting: a qualitative analysis by Tiwonge K Mtande, Carl Lombard, Gonasagrie Nair and Stuart Rennie in Research Ethics
Footnotes
Acknowledgements
We extend our gratitude to study participants and principal investigators of the two studies.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
All articles in Research Ethics are published as open access. There are no submission charges and no Article Processing Charges as these are fully funded by institutions through Knowledge Unlatched, resulting in no direct charge to authors. For more information about Knowledge Unlatched please see here: ![]() The study was supported by funding from the National Institutes of Health: D43TW01511- 01-Advancing Research Ethics Training in Southern Africa (ARESA): Leadership Program.
The study was supported by funding from the National Institutes of Health: D43TW01511- 01-Advancing Research Ethics Training in Southern Africa (ARESA): Leadership Program.
Ethical approval
Health Research Ethics Committee (HREC) at Stellenbosch University & National Committee on Research in Social Sciences and Humanities (NCRSH) in Malawi.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.