
Abstract
There are increasing concerns that research regulatory requirements exceed those required to manage risks, particularly for low- and negligible-risk research projects. In particular, inconsistent documentation requirements across research sites can delay the conduct of multi-site projects. For a one-year, negligible-risk project examining biobank operations conducted at three separate Australian institutions, we found that the researcher time required to meet regulatory requirements was eight times greater than that required for the approved research activity (60 hours versus 7.5 hours respectively). In total, 76 business days (almost four months) were required to obtain the necessary approvals, and site-specific processes required twice as long (52 business days/approximately 10 weeks) as primary Human Research Ethics Committee and Research Governance Office processes (24 business days/ approximately five weeks). We describe the impact of this administrative load on the conduct of a one-year, externally-funded research project, and identify a shared set of application requirements that could be used to streamline and harmonise research governance review of low- and negligible-risk research projects.
Introduction
Research regulation is necessary for institutions to mitigate risks and for researchers to conduct ethically sound research, and is particularly relevant in the face of past research misconduct (Samanta and Samanta, 2005). However, there are growing concerns that regulatory processes are becoming increasingly time-consuming for researchers, without associated improvements in research quality or patient safety (Alberts et al., 2014; Gill and Burnard, 2009; Loscalzo, 2013; Shaw and Barrett, 2006; Shaw et al., 2005, Stein, 2015).
In the case of multi-site human research studies conducted in New South Wales (NSW), Australia, ethics approval for all designated sites is granted by a single certified Human Research Ethics Committee (HREC) (NSW Health, 2010a). Following this, a separate site-specific governance review is conducted by each local Research Governance Office (RGO) for site authorisation (NSW Health, 2010b). Systems that combine single-site ethical review with site-specific research governance review are intended to reduce approval times for multi-site projects, yet unreasonable burdens and delays due to research governance processes have been reported by clinical studies conducted in the UK (Fudge et al., 2010; Galbraith et al., 2006; Kearney et al., 2014; Sandy et al., 2011; Snooks et al., 2012) and in Australia (Barnett et al., 2016; Vaughan et al., 2012). Researchers report inconsistencies in the required documentation between sites (De Smit et al., 2016; Glasziou and Chalmers, 2004; Vaughan et al., 2012) and excessive time spent on repetitive and low-value tasks such as document reformatting (Barnett et al., 2016). In particular, RGO documentation requests for low- and negligible-risk (LNR) research projects can extend beyond those that could be reasonably expected to ensure that study risks are minimised (Galbraith et al., 2006).
Our team secured a one-year research grant for a biobanking project, a component of which involved interviewing biobank staff from three research sites. This study describes the impact of HREC and RGO review on the conduct of this project. Researcher time spent complying with regulatory requirements was compared with researcher time devoted to data collection, and the implications for project efficiency and effectiveness are considered.
Methods and results
Ethics and governance approval at the primary research site and selection of interview sites
According to the National Statement on Ethical Conduct in Human Research, low-risk research involving human subjects only involves foreseeable risks of participant discomfort, whereas negligible-risk research involves foreseeable risks of no more than participant inconvenience (National Health and Medical Research Council, 2007). Participant contact for our project was limited to a 2.5-hour interview with biobank staff, which requested operational information about the facility in which they worked, including salary costs. Interviews were only conducted with biobank staff, and not with any patient biospecimen donors or other patient subjects. The primary HREC considered this project to be of negligible risk, and as such we submitted a low and negligible risk (LNR) application prepared by AR (Figure 1, Table 1). We received HREC and RGO approval in 19 business days (almost four weeks) and five business days (one week) respectively (Figure 1). We then sought Expressions of Interest (EOI) from NSW cancer biobank staff to participate in the interviews, from a cohort of 23 cancer biobanks in NSW. Three biobanks of different sizes (according to the numbers of full-time equivalent staff employed) and at different research sites were selected to participate in the project.
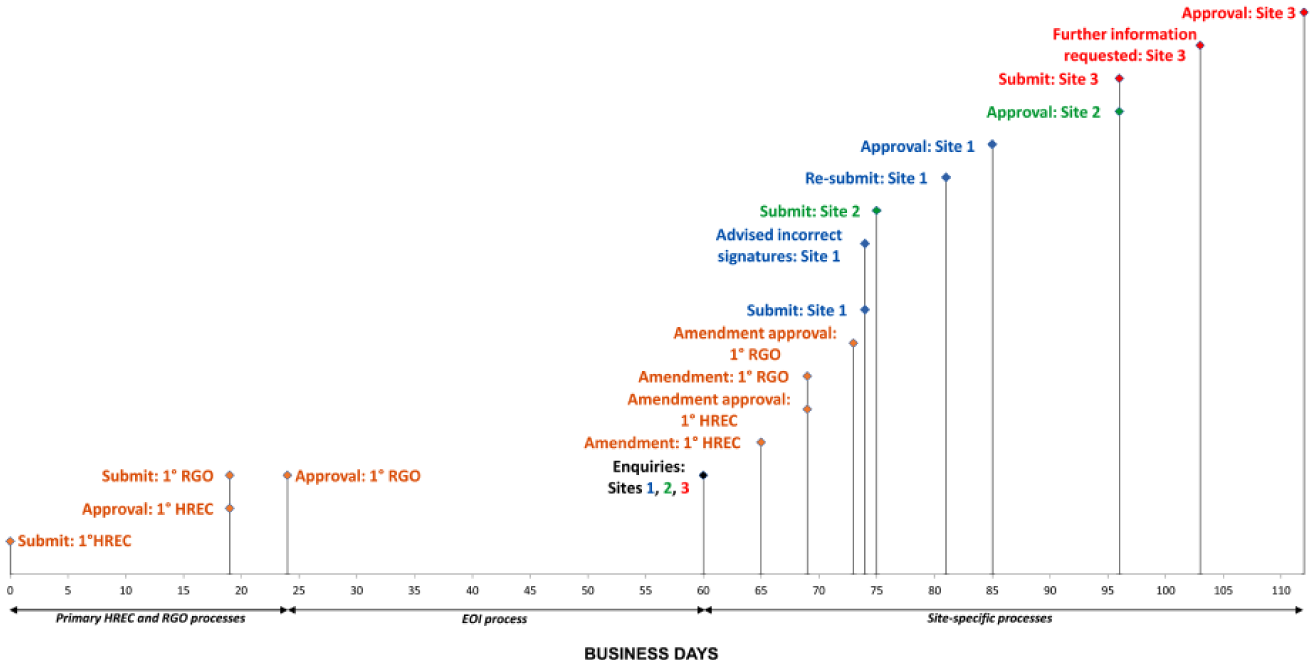
Timeline (x axis, business days, where five business days = one calendar week) showing HREC and RGO approval processes for the primary research site, and RGO approval processes at research sites 1–3. The timeline is divided into three time periods: primary HREC and RGO approvals (days 0–24, orange text), the EOI process (days 24–60) and the site-specific processes (days 60–112, blue, green and red text are for sites 1, 2 and 3, respectively). The time required for the EOI process was not included in subsequent calculations of the time periods required for approvals.
Primary site requirements for low/negligible risk (LNR) research project application.

RGO: Research Governance Office; EOI: Expressions of Interest; HREC: Human Research Ethics Committee; SSA: site-specific assessment.
This correspondence was required to obtain necessary signatures, but was not required to be submitted to the HREC or RGO.
Site-specific assessment (SSA)
The RGOs associated with the three planned interview sites were contacted (Figure 1, day 60). Each RGO reviewed the primary-site-approved HREC application form, research protocol, participant information sheet and interview questions, and confirmed that a LNR Site-specific Assessment (SSA) was required. The LNR SSA is a standard governance application form containing questions about the departments and services involved in the project, the research participants and the project budget. It requires signed endorsement from the study’s principal investigator, co-investigators and the site’s Head of Department (in our case, the person in charge of the biobank staff), as well as the heads of supporting departments (i.e. the person in charge of any department supplying resources to the project). The form is completed online, and can be printed and circulated for wet-ink signatures.
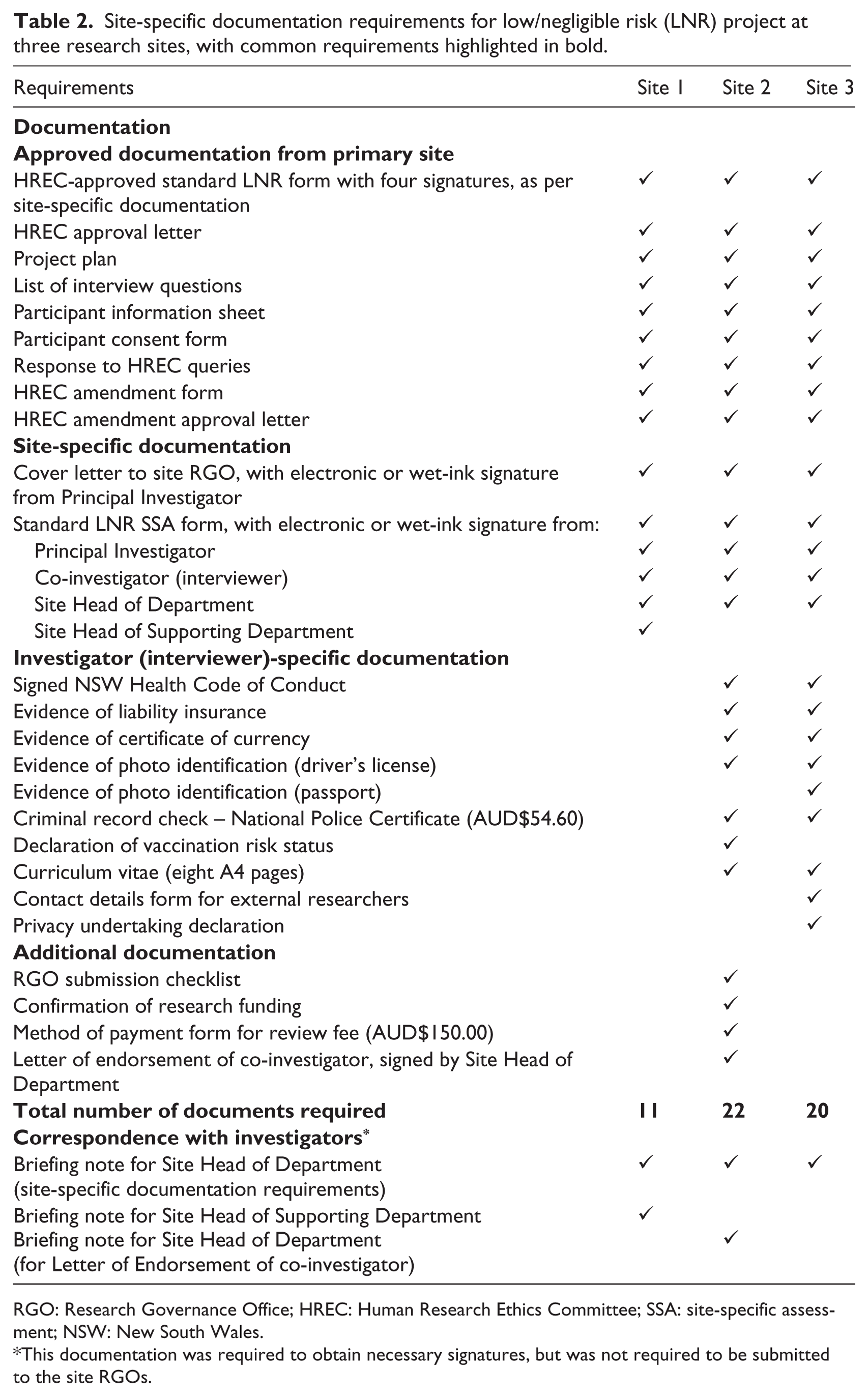
For each research site, SSA submission instructions and requirements (Table 2) were gathered by AR from RGO websites, and by telephone and email. For two sites, 7–9 investigator-specific documents were also required to be supplied for each investigator conducting on-site research (Table 2). Because of time constraints, it was not feasible to obtain these documents from the two planned investigators, and so the task of on-site data collection was assigned to one investigator (RL).
Site-specific documentation requirements for low/negligible risk (LNR) project at three research sites, with common requirements highlighted in bold.
RGO: Research Governance Office; HREC: Human Research Ethics Committee; SSA: site-specific assessment; NSW: New South Wales.
This documentation was required to obtain necessary signatures, but was not required to be submitted to the site RGOs.
Primary site amendment to specify interview sites
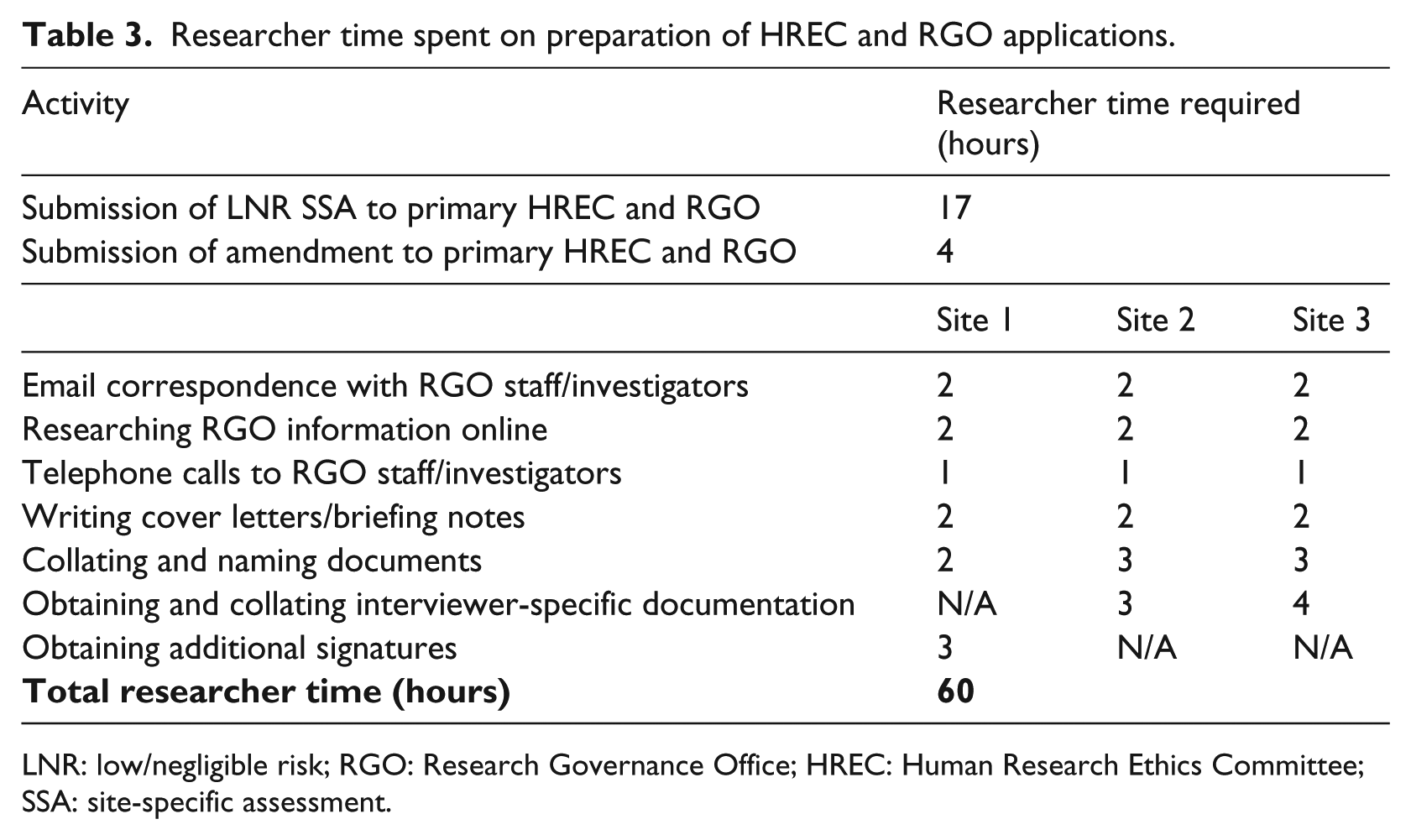
As part of each LNR SSA application, RGOs required evidence of an approved amendment application from the primary site’s HREC and RGO (Table 2). This was for the purpose of specifying the three biobanks selected from the EOI submissions. Preparation for the amendment took four hours of researcher (AR) time (Table 3), and the HREC and RGO amendments were each approved in four business days (almost one week) (Figure 1, days 69–73).
Researcher time spent on preparation of HREC and RGO applications.
LNR: low/negligible risk; RGO: Research Governance Office; HREC: Human Research Ethics Committee; SSA: site-specific assessment.
Site requirements
Almost half (n = 14/29, 48%) of RGO application requirements (11 documents, three signatures) were shared by all three research sites (Table 2), in particular the need to review all documents approved by the primary HREC (Table 1). However, despite our project requiring minimal resources at each site in the form of meeting room access for single interviews, there were also substantial RGO application variations between sites. Site 1 required electronic copies of 11 documents, including four signatures and two briefing notes that described the need for the signatures (Table 2). The LNR SSA application form with briefing notes were posted to the relevant Site 1 staff for their endorsement; however, upon lodging our application to the Site 1 RGO, we were advised that the signed endorsements were incorrect, necessitating a second round of briefing notes, postage and staff signatures. This included an additional signature from the head of the supporting department (Table 2), as the project would utilise institutional resources in the form of a meeting room for the 2.5-hour interview. Obtaining these additional signatures required three hours of researcher time (Table 3) and an eight business day (approximately 1.5 week) delay (Figure 1, days 74–81). Once lodged, the application was approved in four business days (Figure 1, day 85).
The Site 2 RGO required electronic and hard copies of 22 documents, including four signatures and two associated briefing notes (Table 2). A fee of AUD$150 was charged for RGO review (Table 2), and the application was approved in 22 business days (over four weeks) (Figure 1, day 96). Site 3 required electronic and hard copies of 20 documents, including three wet-ink signatures and one associated briefing note (Table 2). Site 3 required more investigator-specific documents than the other sites (Table 2), which delayed the submission of this RGO application (Figure 1). Due to their status as an external investigator, the interviewing investigator RL was also required to present in person to the Site 3 RGO with their previously submitted identification documents. This RGO approved the application in 16 business days (over three weeks) (Figure 1, day 112, pending sighting of RL).
In total, 60 hours of investigator time (Table 3) were required for HREC and RGO processes, and 76 business days (almost four months) were needed to obtain the necessary approvals (Figure 1). Site-specific processes required approximately twice as much time (52 business days, approximately 10 weeks; Figure 1, days 60–112) as primary HREC and RGO approval (24 business days, approximately five weeks; Figure 1, days 0–24). In total, eight times as many researcher hours were spent on regulatory compliance (60 hours, Table 3) as were spent on the approved project interviews (7.5 hours, 2.5 hours per site).
Discussion
This case study of a negligible-risk biobanking research project highlights burdensome and inconsistent research governance requirements across three study sites in NSW, Australia. The level of risk mitigation applied to the site review of this negligible-risk project appeared to be in excess of reasonable requirements. For example, in order to perform a 2.5-hour interview with biobank staff to discuss facility operations, one or more RGOs requested evidence of a national police criminal record check and a declaration of the interviewer’s vaccination risk status. One RGO also required that the interviewer present to their office in person, in order to cross-check their previously submitted proof of identification. These and other requirements resulted in researcher time being diverted from a project that had little associated risk to participants or impact on institutional resources. An ensuing imbalance was created between the amount of time devoted to complying with site-specific regulatory requirements (60 hours), and the time required to complete the site-specific research itself (7.5 hours). Achieving site-specific approvals took approximately twice as long as primary HREC and RGO approvals.
Burdens and delays introduced through complying with research governance requirements have been previously described in the context of clinical or other interventional trials (Barnett et al., 2016; Fudge et al., 2010; Kearney et al., 2014; Snooks et al., 2012) and clinical observational studies (Barnett et al., 2016; Sandy et al., 2011; Vaughan et al., 2012). Most such studies have involved patients as research subjects, although some have involved clinical staff members (Barnett et al., 2016; Galbraith et al., 2006). Just as patients represent vulnerable populations, staff participation in research can present ethical challenges due to the unequal relationship between employers and employees. Nonetheless, it is widely recognised that research undertaken with clinical staff can directly improve patient outcomes (Kim et al., 2009). Staff members therefore provide valuable sources of information for researchers seeking quantitative and qualitative data on workplaces and services, and unnecessary barriers to such research can reduce both transparency and capacity to improve. The present study extends the results of previous clinical studies to suggest that negligible-risk research involving research facility staff is also subject to excessive research governance regulation. Due to the lack of reported data in this area, the extent and consequences of this over-regulation are largely unknown. It is, however, likely that reducing effective access to research scientists as subjects inhibits efforts that might otherwise improve research processes and infrastructure.
A 2009 US survey found that 42% of a researcher’s time is spent addressing regulatory matters, rather than their primary remit (Rockwell, 2009). Diverting research resources to excessive administration has a number of adverse consequences. Firstly, this has the clear potential to alienate funding bodies and to reduce community support for research. Indeed, it could be deemed unethical to spend large proportions of research funds on activities such as RGO applications that are not prioritised in funding applications, and do not improve research quality. Secondly, researcher awareness of challenging RGO application processes may lead to changes to research project design or conduct. In our case, we reduced the number of planned interviewers in response to RGO documentation requirements at two research sites. While this change was minor, other scenarios could include more extensive tailoring of protocols to meet RGO requirements (Al-Shahi Salman et al., 2014). Although researchers have been described as ‘non-voluntary’ clients who must apply to research governance departments in order to carry out research (Fudge et al., 2010), researchers may in some cases have the capacity to avoid and/or target research sites with more/less burdensome RGO requirements. As a result, multi-site research data could be skewed, and research jurisdictions could lose the potential benefits of locally conducted research, such as clinical trials being undertaken within their population. In more extreme cases, research projects may be deferred or not undertaken. As the extent of this problem is likely to be under-reported, study biases in response to RGO requirements may represent an under-recognised contributing factor to research irreproducibility. Finally, excessive regulatory burdens could also reduce research quality by increasing the degree of time fragmentation experienced by researchers. Time fragmentation can inhibit research (Alberts et al., 2014; Gill and Burnard, 2009; Stein, 2015) by requiring researchers to constantly re-engage with complex concepts (Duncan et al., 2015). Thus, excessive regulatory burdens not only reduce the time available for research, but can also reduce the quality of the remaining time, and thus the quality of research that is produced.
To prevent the unnecessary regulation of LNR research, the extents of regulatory assessments should reflect the risk category assigned to research projects (Fudge et al., 2010; Shaw et al., 2005). As risk-benefit balance is dependent upon research type (Glasziou and Chalmers, 2004), the level of RGO scrutiny could be tailored to the proposed project (Shaw and Barrett, 2006), reducing the workloads of both researchers and RGO staff (Fudge et al., 2010). In our case, we noted that the Site 1 RGO required no investigator-specific documentation, suggesting that the extensive documentation required by Sites 2 and 3 was unnecessary (Table 2). The need to provide investigator-specific documentation has been repeatedly identified as a significant burden (Kearney et al., 2014; Sandy et al., 2011), particularly for studies involving many investigators. Instead of extensive investigator-specific documentation (Table 2), a ‘research passport’ that provides a single, authorised documentation check could provide ongoing assurance of an external researcher’s ability to conduct research according to applicable laws and guidelines, while removing the need for researchers to repeatedly supply identification and CVs (Jonker et al., 2011; National Institute for Health Research, 2017; Sandy et al., 2011). The automatic provision of relevant information from the approving HREC to RGOs (Stein, 2015) and/or automated seeding of information in application forms (Al-Shahi Salman et al., 2014) could further streamline research governance applications.
We recognise that variations in local RGO processes may sometimes be necessary in order to account for local circumstances and risks. Nonetheless, as the conduct of our project was uniform across three similar sites, the variations in RGO requirements that we describe support previous results highlighting unnecessary variations in the local implementation of research governance processes (Fudge et al., 2010; Sandy et al., 2011; Snooks et al., 2012). This suggests a lack of awareness as to how process variations affect the conduct of multi-site studies. Contributing factors could include high RGO staff turnover rates which have been reported in different settings (Sandy et al., 2011; Vaughan et al., 2012). Regular intra- and inter-institutional staff training for RGO staff (Pothier and Bredenkamp, 2006) along with mentoring by senior colleagues could reduce unnecessary site-specific process variations, as well as provide perspectives on risk mitigation. This could be strengthened by top-down approaches from federal or state-wide government institutions to actively incentivise harmonisation (Snooks et al., 2012; Stein, 2015), noting that government-imposed targets have been reported to improve research-governance-approval time frames in the UK (Kearney et al., 2014). Australia’s National Health and Medical Research Council has recently released guidelines that aim to streamline the governance review of clinical trials (National Health and Medical Research Council, 2016). A similar set of guidelines for LNR projects could be developed to ensure the standardisation and harmonisation of RGO review. In particular, the site-specific application requirements that were common to the three research sites (Table 2) could be broadly adopted for RGO review of LNR projects.
In summary, tailored and harmonised research governance processes would reduce the workloads of both researchers and governance staff (Kim et al., 2009; Stein, 2015). For researchers, this would result in more time and energy to focus on research, leading to better quality research through an improved capacity to engage with complex ideas, and fewer biases introduced through, for example, the selection of research sites based on administrative requirements. Reduced focus on LNR research projects would also allow RGOs to devote more resources to higher-risk projects (Fudge et al., 2010; Kim et al., 2009). We propose that the common set of RGO application requirements identified in this study could help to rationalise and harmonise the review of LNR research projects, to the benefit of researchers, administrators, funding organisations and, ultimately, the general public.
Footnotes
Funding
All articles in Research Ethics are published as open access. There are no submission charges and no Article Processing Charges as these are fully funded by institutions through Knowledge Unlatched, resulting in no direct charge to authors. For more information about Knowledge Unlatched please see here: ![]() .
.