
Abstract
Pragmatic clinical trials (PCTs) are a relatively new methodological approach to the execution of clinical research that can increase research efficiency and provide access to unique data. Some have suggested that the costs and delays associated with obtaining informed consent could make PCTs difficult or even impossible to execute. Alternative consent models have been proposed, some of which lower standards of disclosure, delay consent, or waive it altogether. We analyze the permissibility of changes to informed consent in the context of Canadian research ethics policies, legislation, common law, professional codes of ethics, and professional standards of practice. We find that Canadian law and policy relating to informed consent clearly applies to any clinician who might be involved in a PCT. In addition, existing consent norms seem unable to accommodate alternative consent models for pragmatic research if such models would involve lowering the standard of disclosure. The strong emphasis on the primacy of individual rights that exist in law and in research ethics norms cannot easily coexist with strategies that involve either waiver of consent requirements or the provision of incomplete information about the research prior to enrolment. If Canadian policy-makers wish to create the regulatory flexibility necessary to accommodate altered consent and disclosure, it is likely this will require the alteration of existing health information legislation, national research ethics policy, and professional standards.
Introduction
Pragmatic clinical trials (PCTs) are examples of relatively new methodological approaches to the execution of clinical research. In general, PCTs focus on the collection of data in the real world clinical context (Buyse, 1993; Cortese, 2015; Glasgow, 2013; Rowbotham et al., 2013; Wahba et al., 2010; Williams et al., 2015). Though approaches to PCT design can vary greatly, common pragmatic features include: (i) a focus on public health outcomes; (ii) use of multiple heterogeneous settings to conduct research; (iii) minimization of exclusion criteria for participants: and (iv) comparison of existing alternative interventions that often fit a typical standard of care, rather than the use of placebo (Williams et al., 2015). Blinding is only sometimes attempted (Lowe et al., 2011), as the approach is meant to reflect real world usage. Uncontrolled variables are not always avoided in the same way they are in classical RCTs. Flexibility, efficiency, and cost relative to sample size can be greatly improved in PCTs as compared to traditional RCTs. Large sample sizes, decreased need for investigator training, easier data collection and a faster overall process are all, at least in part, achieved in PCTs through the integration of research into existing clinical workflow (Albright et al., 2013; Alford, 2006; Ali et al., 2015; Larson et al., 2016; Lu et al., 2014). PCTs can yield fewer false negatives (Buyse, 1993), and they often prioritize external validity in the context of routine clinical practice conditions (Chalkidou et al., 2012; Delitto, 2016). PCTs are often large in scale, and clinical care resources are utilized in their implementation (Concannon et al., 2014).
Given the growing interest in these forms of research, it is important to consider the legal and ethical issues associated with their deployment. The requirements surrounding the process of informed consent, as well as the tension between the norms associated with the conduct of research and the provision of healthcare can make the execution of a PCT particularly difficult (Buyse, 1993; Gandhi et al., 2015; Ishak et al., 2011; Saag et al., 2012). Indeed, some proponents of PCTs claim that existing standards for informed consent are incompatible with the execution of scientifically sound PCTs, as they introduce biases in data and can render certain study designs impracticable (Anderson et al., 2015; Sugarman and Califf, 2014; Truog et al., 1999). In addition, there is concern about the cost and logistics of obtaining consent in this context (Sugarman and Califf, 2014). Consequently, concepts of altered consent (consent that does not satisfy all the typical criteria of informed consent) and delayed consent (consent collected after or partway through participation in research) have been suggested. Examples of proposed alterations include waiver of consent, broadcast notification of PCT in prominent locations, integration of clinical and research consent, short-form consent, and electronic consent (McKinney et al., 2015; Wendler, 2015).
PCTs have already been undertaken in various jurisdictions, including Canada. Many of these trials include a standard approach to obtaining informed consent, but others utilize an altered consent process (Dumbleton et al., 2015; Fröbert et al., 2013; Goering et al., 2011; Selak et al., 2016). The legal permissibility of alterations to or waivers of consent can vary by jurisdiction, though overarching national and international ethics guidelines also inform the applicable regulatory frameworks. In this article, we review and analyze the relevant sources of Canadian law and research ethics policy, using Alberta as a provincial example, in order to assess the feasibility and permissibility of PCTs under the existing legal regime and relevant research ethics policy. Following this analysis, we consider the future of PCTs given the current regulatory environment.
The consent problem
In Canada, consent law requires clinicians to provide patients with anything a reasonable person in the patients’ position would want to know (Reibl v Hughes, 1980). The courts have determined this to be a relatively expansive obligation and, in the context of research, it has been characterized as ‘the most exacting duty possible’ (Picard and Robertson, 2007). This means that clinicians need to disclose all relevant risks. In general, Canadian research ethics policy has mirrored these legal standards, requiring disclosure of, among other things, all information relevant to participation in research, all reasonably foreseeable risks and potential benefits, and information about the possibility of commercialization of research results (CIHR, 2014). Although there are exceptions to the obligation to get fully informed consent for research (as we will discuss below), in the clinical setting, fully informed consent has long been the legal and ethical norm – and this can create significant policy challenges for those interested in doing PCTs.
To help understand the consent challenge, consider this hypothetical example of a PCT design for testing comparative effectiveness. A researcher wishes to determine which of two drugs, both of which satisfy existing clinical standard of care, is superior. There is no clear indication that one drug is better than the other. Given these facts, the research would satisfy most definitions of clinical equipoise (genuine uncertainty as to which intervention is superior) and minimal risk (possible harms of the research are no greater in magnitude and probability than those encountered by participants in aspects of everyday life that relate to the research) (CIHR, 2014). In order to generate a large sample size, the methodology involves recruiting patients in both hospitals and clinics. The participating health professionals would randomly assign one of the drugs to new patients, and would collect and anonymize patient data on outcomes. Written informed consent would cost a significant amount per person and it is possible it could reduce participation. Therefore, the researcher applies to the relevant authority, such as a research ethics board (REB), for approval (for funding purposes, which is distinctly different than a legal right) to waive the requirement to obtain informed consent. If this waiver is granted, participants would not necessarily know they were involved in research or that their drug had been randomly assigned until after it was given to them. Of course, they would still need to be informed and consent to the medical treatment, likely orally.
Heddle et al. undertook a pragmatic randomized controlled pilot trial that appears to be very similar to the hypothetical described above, and published their results in 2012. The study compared the mortality rate of patients needing a blood transfusion when they received either the freshest available blood or the oldest available blood that met existing limits of standard of care, and the researchers received permission from a Canadian REB to waive consent (Heddle et al., 2012). The blood was thus assigned randomly without informed consent to participate in research. The researchers wrote that without a consent waiver such a study ‘would not be feasible, because it would not be possible to recruit the majority of patients who require a blood transfusion’ (Heddle et al., 2012).
The primary legal and ethical concerns with PCTs arise from variations from existing norms of informed consent (Califf and Sugarman, 2015; Caulfield and Ries, 2003; Kalkman et al., 2015). As noted above, although exceptions exist, the requirement of specific informed consent is, in general, the accepted approach to obtaining consent in the context of clinical research. Much has been said about the challenges of research using broad consent (i.e. consent that is sought once and applies to a wide range of known and unknown future research) (Master et al., 2012). Although this approach is often used in the context of, for example, biobank studies, it remains controversial, as it is a significant departure from existing norms (Nanibaa’A et al., 2015). The reasons broad consent is used in biobanking are similar to the issues with PCTs. Biobanks often involve thousands of participants, and the data may be used for numerous different studies involving a large number of researchers. If specific consent was required for each study, it could render a biobank project infeasible because of labor requirements and cost. As such, the research community has pushed for alterations to and waivers of consent, though there remains little consensus as to whether these are legally appropriate (Hodge and Gostin, 2017; Master et al., 2012). Researchers undertaking PCTs face similar challenges related to efficiency. However, biobank research usually involves only the collection of tissue and information; it is not, in general, clinical research involving a specific intervention. In addition, PCTs take place in a clinical setting, with all the concomitant legal and ethical obligations that flow from a physician/patient relationship, i.e. those that mandate prioritizing the best interests of individual patients above all other interests. As such, those undertaking PCTs face even more formidable research ethics questions: is informed consent always necessary in clinical research, and to what extent must it be comprehensive, in the traditional sense, when clinical research overlaps with medical care?
Alterations to and waivers of the consent process have the potential to affect the willingness of patients/participants and institutions to participate in PCTs, and may affect whether and how research results are disseminated (Coronado et al., 2016; Whicher et al., 2015b). For example, for all publications following the International Committee of Medical Journal Editors’ recommendations, authors must satisfy the editors that appropriate consent norms have been satisfied (ICMJE, 2017). As such, if researchers fail to provide evidence that appropriate consent was obtained for clinical research, this will have a direct impact on the ability to publish results.
In addition, there are reasons to believe that altering the consent process may impact public support. Multiple studies on the issue of consent in pragmatic research indicate that the majority of the public favors specific informed consent in the context of a PCT (Nayak et al., 2015; Weir et al., 2014; Whicher et al., 2015a). Indeed, our own Alberta-based survey of 1211 people found that a striking 93.2 percent of respondents believed doctors should obtain consent before enrolling patients in a trial that includes random assignment between two drug treatments (see Text Box 1). Therefore, PCTs using altered, waived, or delayed consent may be perceived by the public as involving an unacceptable erosion of patient rights (Caulfield and Ries, 2003).
Survey B on Medical Consent and Tissue Donation (undertaken in the province of Alberta).
In July 2015, the University of Alberta Population Research Laboratory conducted a randomized phone survey to collect information about public attitudes toward issues of medical consent. The questions, as well as the relevant response data, are below. Before asking the questions, the interviewer gave the following context:
‘Imagine that you need to be treated with a cholesterol lowering drug. There are several types of these drugs, and all are similar to one another in terms of price, size and composition. All of the drugs lower cholesterol and all have been shown to prevent heart attacks. Despite their similarities, it is possible that there are slight differences between the efficiency, effectiveness and safety of these drugs.
Some doctors may prefer to prescribe Drug A and others may prefer to prescribe Drug B, but there is no clear evidence which drug may be better than the other.
As part of a research project to study the drugs and determine which one may be better, your doctor plans on prescribing half of his patients Drug A and the other half Drug B. He will determine which patients get which drug randomly like flipping a coin.’
Q1. As one of your doctor’s patients, how comfortable are you with the idea that the drug you are prescribed will be determined by random like flipping a coin?
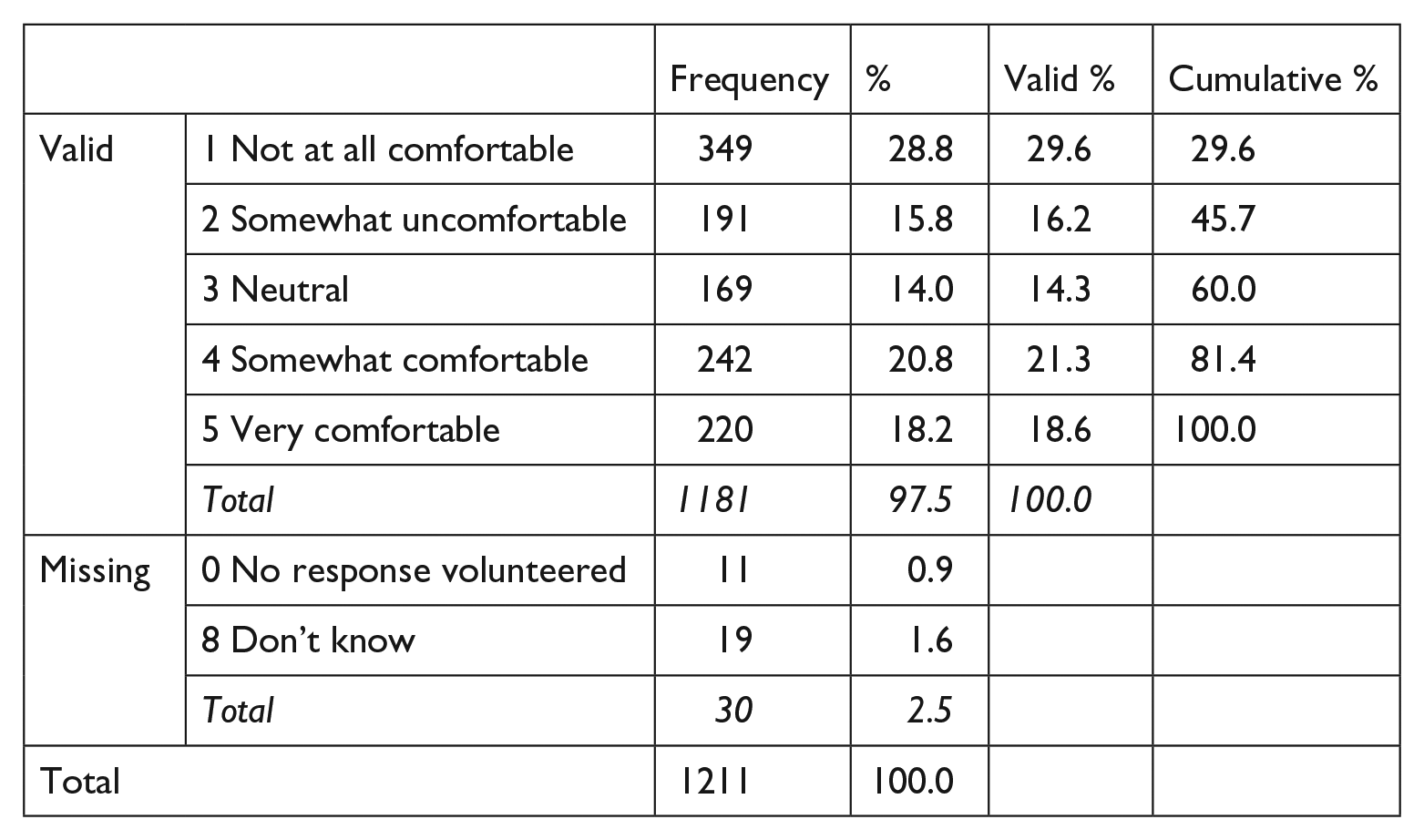
Q2. Do you think that the doctor should obtain consent from his patients to be part of the research project before he determines whether the patient should randomly be given Drug A or Drug B?
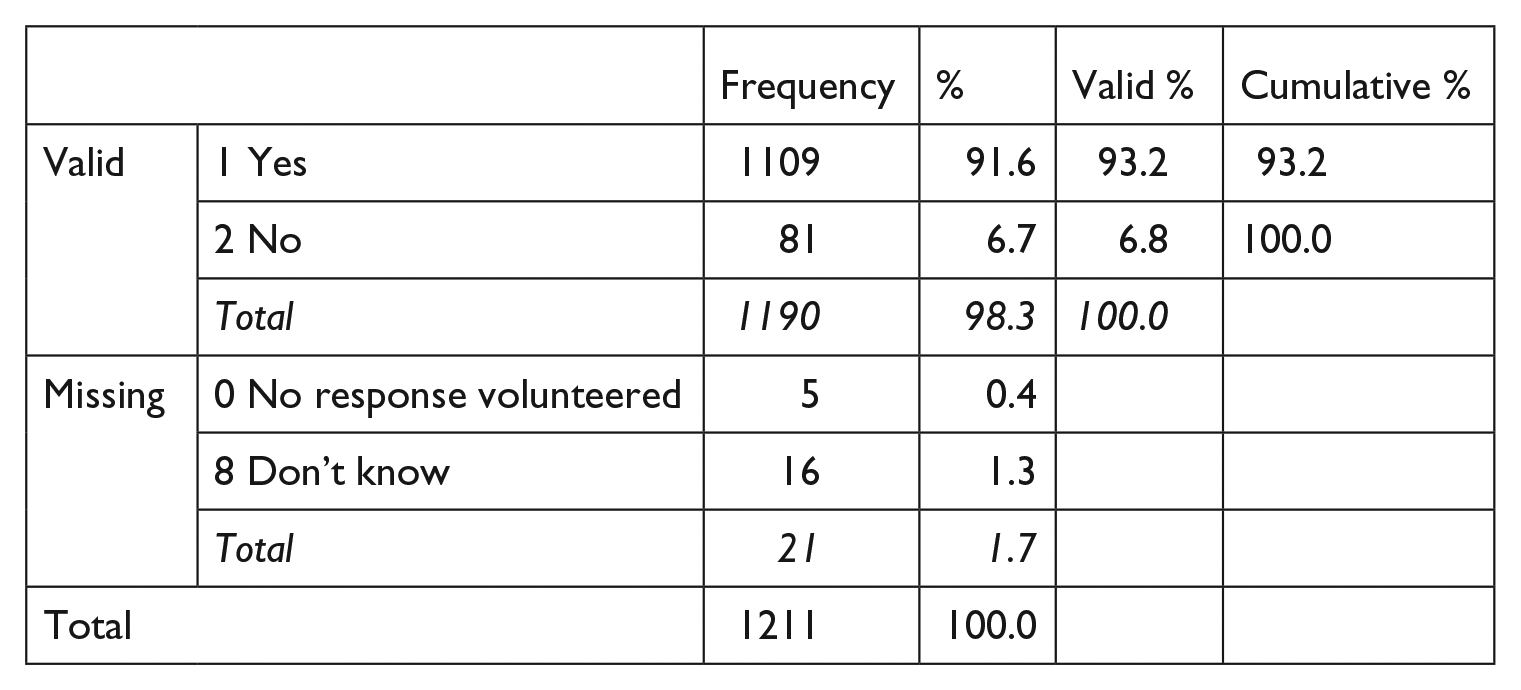
Data collected on the characteristics of the respondents indicated that they were more likely to be white, above average in household income, married, homeowners, over age 45, and have a lower level of education than a university bachelor’s degree.
In some situations, the use of PCTs may also have an adverse impact on the clinical environment. Research has shown, for example, that PCTs may have a negative impact on the provider–patient relationship (Whicher et al., 2015a). If patients come to learn that their doctors can and do withhold information about treatment decisions for the purposes of research, there is the possibility that trust and openness will be diminished.
Proposed alternatives to informed consent
Applicable law and research ethics policy
(i) The Tri-Council Policy Statement
In Canada, the Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans (TCPS2) sets the national standard for clinical research ethics (CIHR, 2014). Institutions and researchers must agree to comply with the TCPS2 in order receive federal funding from any of the three primary research agencies (CIHR, 2014). Many sections are relevant to the PCT consent issue.
Chapter 1 outlines the policy’s foundational principles, including ‘respect for persons’, ‘concern for welfare’, and ‘justice’ (CIHR, 2014). Respect for persons ‘reflects the commitment that participation in research … should be a matter of choice and that, to be meaningful, the choice must be informed’ (CIHR, 2014). Altered or waived consent prioritizes research interests rather than those of the research participant.
Chapter 3 of the TCPS2 outlines standards for informed consent, noting that it must precede collection of or access to research data, and must be voluntary, informed, and ongoing (CIHR, 2014). In the context of a PCT, neither delayed consent nor waiver of consent models can ensure that participants are informed and participate voluntarily. Article 3.2 states that prospective participants shall receive ‘full disclosure of all information necessary for making an informed decision to participate in a research project’ (CIHR, 2014). This includes, among other things, the research purpose, the details of the procedure, all reasonably foreseeable risks and potential benefits, the identity of the researcher(s) and funder(s)/sponsor(s), information concerning the possibility of commercialization of research findings, and an assurance that there is no obligation to participate (CIHR, 2014). Altered or waived consent models limit participant rights to make decisions as to what kind of commercial endeavors to support.
Article 3.5 states that ‘research shall begin only after the participants, or their authorized third parties, have provided their consent’ (CIHR, 2014). This precludes delayed consent models, though exceptions exist when approved by an REB in accordance with Articles 3.7 (consent alterations) and 3.8 (emergency research) (CIHR, 2014). Article 3.7A allows an REB to approve a clearly defined alteration to consent where the research involves no more than minimal risk, the alteration is unlikely to adversely impact the welfare of participants, and research is impossible or impracticable if prior consent of participants is required (CIHR, 2014).
For research to be impracticable, it must be ‘incapable of being put into practice due to a degree of hardship or onerousness that jeopardizes the conduct of the research’ (CIHR, 2014). The example given is psychological research to see whether people would return a wallet that was dropped in front of them (CIHR, 2014). Indeed, the text states that ‘if the aims of the research can be achieved with a design that allows for full – or fuller – prior disclosure (in accordance with Articles 3.1–3.5), then that design must be adopted’ (CIHR, 2014). Proponents of reduced consent obligations would claim that the prohibitive costs of informed consent, the related effect of reducing the number of participants in research, and introduction of consent bias make pragmatic research impracticable (Sugarman and Califf, 2014; Truog et al., 1999). REBs need to consider whether decreased cost efficiency and sample size truly constitute impracticability as it is defined in the TCPS2. We suggest that the wording seems to indicate that it does not. With PCTs, consent is possible, but expensive. And although it may impact recruitment, impacts of this type do not affect the core ability to execute the research, as with the psychological study example provided in the TCPS2. In other words, research is still possible with the provision of consent, but it is expensive and may result in a less than ideal data set.
Chapter 11 speaks to key requirements in clinical trial design and execution, including establishing clinical equipoise, minimizing risk, and maintaining the dual role of clinician-researchers (CIHR, 2014). It notes that clinician-researchers in a dual role can problematically conflate their clinical practice with clinical trial research, and states that ‘to preserve the trust … researchers should take all necessary measures to separate their role as researcher from their role as clinician’ (CIHR, 2014). This section of the TCPS2, again, suggests that in the context of PCTs, REBs should be cautious about moving away from a robust approach to consent. Preservation of trust could prove difficult if informed consent requirements are eroded.
(ii) Other research ethics standards
There are a number of other research ethics guidelines relevant to the issues associated with PCTs. Although some of these are not technically binding on researchers in Canada, they do inform norms of acceptability among researchers and research ethics bodies. Notably, the Declaration of Helsinki is a foundational international research ethics document, and one of its general principles is that ‘While the primary purpose of medical research is to generate new knowledge, this goal can never take precedence over the rights and interests of individual research subjects’ (World Medical Association, 2013). Alterations to informed consent procedures that curtail patient rights for the purpose of research efficiency have the potential to conflict with this foundational norm.
The Canadian Medical Association’s Code of Ethics is relevant to PCTs because clinicians administer PCTs alongside standard clinical care. The Code largely mirrors many principles found in the TCPS2 (Canadian Medical Association, 2004). Listed clinician responsibilities include providing patients with the information needed to make informed decisions about medical care, communicating in a manner so as to be understood, respecting the right of a competent patient to accept or reject any medical care, protecting personal health information of patients, obtaining consent before disclosing patient information to third parties, and obtaining full informed consent from potential subjects before proceeding with any study (Canadian Medical Association, 2004). These responsibilities effectively prohibit clinicians from adopting changes to informed consent that erode disclosure requirements.
At the provincial level, the various self-regulating medical colleges can impact the permissibility of altered consent in research. For example, the College of Physicians and Surgeons of Alberta’s Standard of Practice for Human Health Research states that a regulated member participating in human health research must ‘disclose to patients that the study has been reviewed by an ethics board and relevant conditions imposed’ (College of Physicians & Surgeons of Alberta, 2015). Standards of practice are enforceable under the Alberta Health Professions Act, RSA 2000, c H-7, making this a binding obligation. This clause seems to preclude study designs where researchers do not inform participants about their participation, though it may leave room for alterations to consent.
(iii) Canadian health information legislation
In Canada, there is plenty of legislation covering the use of personal information, but little is of relevance to the consent issue present in PCTs. Federally, the Privacy Act, RSC 1985, c P-21 only applies to government institutions, and the Personal Information Protection and Electronic Documents Act, SC 2000, c 5 only applies to research if it qualifies as a commercial activity, such as marketing research.
In general, the provinces are left to regulate the collection, use, and disclosure of health information for health research. In the Alberta example, the Health Information Act, RSA 200, c H-5 (HIA) governs. However, the HIA speaks only to the alteration of consent for the collection, disclosure and use of information. It does not address the alteration of consent to participate in a clinical trial and be subjected to an intervention in accordance with a research protocol. As such, it is unlikely that provisions in the HIA that allow altered consent for the collection and use of health information with REB approval are applicable to the core consent issues with PCTs (HIA, ss. 19, 26, 27(1), 50).
(iv) Canadian common law
In Ciarlariello v Schacter [1993] 2 S.C.R. 119 (at 135), the Supreme Court of Canada (SCC) explicitly notes that Canadian consent law largely flows from application of the ethical principles of autonomy and physical integrity. Informed consent requires the disclosure of relevant risks. In Reibl v Hughes [1980] 2 S.C.R. 880, the SCC held that physicians must, as part of the consent process, disclose anything a reasonable person in the patient’s position would want to know.
In Halushka v University of Saskatchewan (1965) 53 D.L.R. (2d) 436, the court held that, owing to the lack of intended therapeutic benefit for participants, the threshold for informed consent is higher for medical research than it is for ‘mere medical treatment’. Indeed, as noted by Picard and Robertson (2007), consent in the medical research setting should be viewed as one of the most exacting duties possible. As per Halushka, participants are entitled to ‘full and frank disclosure’ of all relevant facts, probabilities, and opinions a reasonable person might be expected to consider before giving consent, regardless of whether minor disclosures might cause unnecessary worry.
In addition, in Canada, physician–patient relationships are considered fiduciary in nature (McInerney v MacDonald [1992] 2 SCR 138; Norberg v Wynrib [1992] 2 SCR 226). Because clinical researchers are acting as physicians to patient-participants, they too must uphold fiduciary obligations. Canadian common law requires that a fiduciary (i.e. physician-researcher) act honestly, in good faith, and strictly in the best interests of a beneficiary (i.e. patient-participant) (Canson Enterprises Ltd. v Boughton & Co. [1991] 3 SCR 534). Prioritizing research over the disclosure of relevant treatment information conflicts with these duties. Therefore, as information that could affect the patient’s health, autonomy, privacy, or other ‘vital’ or ‘practical’ interests arises, such as the potential for modification to or randomization of his/her treatment in accordance with research, fiduciary law dictates that disclosure is mandatory (Canson Enterprises Ltd. v Boughton & Co. [1991] 3 SCR 534).
Given this jurisprudence, the existing Canadian law of consent, which clearly applies to any clinician who might be involved in a PCT, seems unable to accommodate alterations to or waivers of consent for pragmatic research if such models would involve lowering the standard of disclosure.
Discussion: Current incompatibilities
The review of law and policy above illustrates some important findings. First, key bioethics policies that allow for deviations from the standard procedures for informed consent only allow such deviations in limited circumstances. Sometimes these circumstances are equivocally defined, in order to preserve the discretionary power of REBs. However, in the case of the TCPS2, one can reasonably conclude from the definitions and examples given in the text that increased cost and difficulty in recruiting participants are not sufficient to render a study ‘impracticable’, and upon accepting this, the justifications of many potential PCTs for altering or waiving consent are rendered invalid (CIHR, 2014). Second, clinicians’ ethical obligations effectively prohibit them from undertaking any behavior that prioritizes research interests over patients’ interests. Third, binding clinician standards of practice can effectively prohibit the use of delayed consent or waiver of consent. Finally, the common law’s delineation of disclosure requirements for informed consent does not contemplate or authorize any exceptions for the purpose of furthering research interests, indicating a disconnect between the law and existing research ethics standards.
Indeed, the strong emphasis on the primacy of individual rights that exists in law and in research ethics cannot easily coexist with strategies that involve either waiver of consent requirements or the provision of incomplete information about the research prior to enrolment. This seems particularly so when research is occurring in a clinical setting, a context where physicians have a clear obligation to place the interests of their patients first. Given this reality, designing PCTs that do not adhere to these norms will prove challenging. Although pragmatic trials certainly vary in the degree to which consent is an issue (Ali et al., 2016), at the current time there seems little room to accommodate the ‘no consent’ approach while maintaining the principle of respect for persons (Kim and Miller, 2016). From a research perspective, there are many potential advantages to PCTs. In general, they are quicker and more efficient, and provide unique clinical data. If Canadian policy-makers wish to create the regulatory flexibility necessary to accommodate these types of methods, it is likely this will require the alteration of existing health information legislation, national research ethics policy, and professional standards.
Footnotes
Acknowledgements
This manuscript was completed as part of the Interdisciplinary Chronic Disease Collaboration (ICDC). The ICDC is funded by the Alberta Innovates Health Solutions CRIO Team Grants Program. The authors would like to thank Marcello Tonelli and Maeghan Toews for designing the survey questions mentioned in Text Box 1. They would also like to thank Ubaka Ogbogu, Robyn Hyde-Lay, and Alessandro Marcon for their ideas and suggestions. Finally, the authors would like to thank all participants in the Health Law Institute workshop titled ‘Emerging Research Methods and the Consent Challenge’, held 25–27 September 2016 in Banff, Alberta.
Declaration of conflicting interests
The authors declare that there are no conflicts of interest.
Funding
All articles in Research Ethics are published as open access. There are no submission charges and no Article Processing Charges as these are fully funded by institutions through Knowledge Unlatched, resulting in no direct charge to authors. For more information about Knowledge Unlatched please see here: ![]() .
.