
Abstract
Background:
Recurrent vulvovaginal candidiasis management primarily entails azole therapy used as required or as an extended daily or weekly maintenance therapy for 6 months or more. Unfortunately, relapse within 3–6 months of ceasing maintenance therapy is experienced for more than half the patients, for whom indefinite treatment is required.
Objectives:
To explore the feasibility of trial design examining a prophylaxis treatment to prevent recurrent vulvovaginal candidiasis symptomatic episodes and reduce adverse effects.
Study Design:
A double-blinded randomized controlled feasibility trial was conducted in Australia. Women with recurrent vulvovaginal candidiasis were enrolled.
Methods:
An intravaginal prophylaxis application of lactic acid and acetic acid (Intravaginal Combination Therapy of Acetic and Lactic Acid) was compared with placebo. Primary outcomes comprised recruitment and retention, compliance to study medications and study assessments. Secondary outcomes included the reduction of symptomatic recurrence over the trial period and the acceptability, satisfaction, safety and tolerability of the intervention. The feasibility of quality-of-life measures was also explored.
Results:
Fifteen participants were enrolled and randomized (active = 9, placebo = 6). Consent rate was 23.4%. Eight participants were lost to follow-up (active = 5, placebo = 3). Forty-seven per cent of participants (n = 7) were 100% compliant with the intervention, six of which completed the trial with good assessment compliance. The blinding process was effective. The study demonstrated a reduction in relapse in both active and placebo groups with only four participants across both groups reporting symptomatic episodes while enrolled. The intervention demonstrated good tolerability. Quality-of-life data showed minimal variance with a high quality-of-life measure.
Conclusion:
This trial assesses the feasibility of conducting a large-scale study exploring the efficacy of the Intravaginal Combination Therapy of Acetic and Lactic Acid intravaginal intervention and hints on the importance of psychological support through appropriate disease-specific communication and clinical attention. Consideration of the reported recruitment challenges, the inclusion of suitable quality-of-life measures and digital data collection is warranted for adaptation to a fully powered trial.
Keywords
Introduction
Up to 10% of the adult female population experience recurrent vulvovaginal candidiasis (RVVC), a disorder with severe physical and psychological impacts despite pharmacotherapeutic options such as topical and oral antifungal azole medicines. 1 Azole therapy may be used as required or as extended daily or weekly maintenance therapy for 6 months or more.2,3 These maintenance regimes often utilize fluconazole for its better safety profile. A Cochrane Review showed that 57% of people with RVVC experience relapse within 3–6 months of ceasing maintenance therapy. 4 Consequently, for some patients, indefinite treatment is required. 5 There is a clinical need for treatment alternatives that can reduce RVVC symptomatic episodes and be used long-term without adverse effects. In addition, there is a lack of evidence-based treatment options for pregnancy, conception and pre-existing health issues, where extended antifungal drug treatment is not clinically appropriate. There may also be a personal preference against long-term drug therapy.
Topical acids such as lactic acid (LA) and acetic acid (AcA) are used as over-the-counter (OTC) medicines in preventing and treating recurrent vaginal infections. Despite in vitro evidence that indicates AcA can decrease the growth of resistant strains of Candida albicans and non-albicans strains such as Candida glabrata, there is a sparsity of research exploring the use of AcA in managing RVVC.6 –8 Non-albicans Candida strains are estimated to account for between 11% and 80% of recurrent and resistant infections, dependent on patient-specific factors.6 –8 The fungicidal in vitro effects of AcA are associated with an influx of AcA into the yeast cell, contributing to the yeast cell’s apoptosis and suggesting that AcA may effectively reduce recurrent infection in RVVC sufferers.6,7 Human trials report favourably on LA’s ability to address the vaginal microbiome dysbiosis associated with bacterial vaginosis (BV). Despite this, there is a lack of high-quality evidence supporting LA’s use for vaginal microbiota modulation. 9 LA’s effects have not been evaluated in human fungal infections; however, literature exploring the bacterial microbiome in RVVC suggests that beneficial microbial shifts and pH regulation could be advantageous for some RVVC cases. 10 Case studies and susceptibility testing utilizing LA have reported reduced local inflammation, which may decrease immune hypersensitization. 11
An AcA and LA combination therapy may provide synergistic ways of reducing Candida recurrence by impacting other microbes such as protozoa, bacteria and fungi.12 –15 Intravaginal applications of 3% AcA and 2% LA are considered safe, with 5% LA used in commercial vaginal health products and clinical studies to treat bacterial vaginal infections such as BV. 16 AcA is considered non-hazardous at concentrations up to 5%. 17 A clinical trial using AcA vaginally for acidification demonstrated 3% AcA to be safe and well tolerated. 18
This study assesses whether a double-blind randomized controlled feasibility trial of intravaginal prophylaxis management in RVVC individuals is appropriate in design and feasibility. Specifically, it investigated (1) recruitment and retention, (2) adherence to trial procedures and intervention protocol and (3) acceptability and suitability of outcome measures and trial procedures. In addition, the trial explored if ICTALA (Intravaginal Combination Therapy of Acetic and Lactic Acid) is an effective and well-tolerated intervention for preventing RVVC symptomatic episodes.
Methods
Study design
A double-blind randomized controlled feasibility trial was conducted via telehealth in Australia, evaluating a trial design which compared an intravaginal prophylaxis application of LA and AcA to placebo. Trial recruitment began in January 2021 and ended in March 2022, with all enrolled participants completing data collection by November 2022. The trial was registered with the Australian New Zealand Clinical Trial Registry (www.anzctr.org.au; ACTRN 12620001084976). The full trial protocol is available from authors on request.
Figure 1 outlines the trial design for a participant enrolled in the 9-month study. Trial baseline and Investigational Medicinal Product (IMP) application windows aligned with the participants’ menstrual cycle or menstrual cycle equivalent if using an oral contraceptive pill (OCP). The trial utilized 1 week of intravaginal treatment repeated monthly for 6 months (aligned with cycle day 10 (±2 days)), followed by a 3-month observation phase. Participants completed paper-based diaries and health assessments fortnightly throughout the 9-month trial period. The study design and subsequent reporting utilized the CONSORT 2010 Checklist extension for Feasibility Trials. 19

Study design.
Study participants
People with RVVC were recruited via social media, HealthMatch (an online clinical trial recruitment platform), University internal emails (to staff and students), local pharmacies, and general medical practices between October 2020 and March 2022.
After initial phone prescreening, an information pack and consent documents were emailed to potential participants with encouragement to ask clarifying questions. After providing electronic informed consent, potential participants completed home eligibility screening tests which consisted of a home pregnancy test, Savvy yeast check, Sobel score assessment and vaginal pH test. Eligible participants were enrolled, randomized, and allocated to either the placebo or active IMP group.
Premenopausal women in good health with a confirmed history of RVVC defined as ⩾4 episodes of vulvovaginal candidiasis in 12 months documented in patient medical records and case history, including at least one positive vaginal culture pathology report, were invited to participate. Patients were eligible only if they were not in an acute symptomatic state (confirmed by a negative Savvy yeast check 20 ) and indicated a Sobel score less than three 21 at baseline, as the intervention was designed to prevent symptomatic periods, not treat acute flares. The Sobel score 21 assesses symptoms such as itching, redness, burning and oedema was scored semi-quantitatively as 0 (absent), 1 (mild), 2 (moderate) or 3 (severe) to assess symptom severity. A composite score of ⩾3 was seen as a symptomatic attack. 21
Excluded from participation were women with intrauterine and intravaginal devices (IUD – hormonal or coil, ring pessary) due to potential interaction with the intervention and known impacts of inserted devices on vaginal microbes and potential infection risk,22,23 and women using any medication that addressed hormones, including progesterone-only contraceptives (POCs), hormonal therapy (except the combined OCP), antiestrogens, aromatase inhibitors or those taking β-hydroxy β-methylglutaryl-CoA (HMG-CoA) reductase inhibitors, antiretroviral or immunosuppressant medication. Moreover, women with significant chronic disease or illness except medicated Hashimotos and latent herpes simplex virus type 1 and 2 (HSV1, HSV2) were not eligible to participate. Participants with a known allergy to any of the ingredients in the IMP formulation, confirmed as pregnant via urinalysis at screening and those not willing to use effective methods of contraception for the trial duration were also ineligible for the study. Ongoing antimicrobial or anti-inflammatory medications also prohibited enrolment, as did taking study permitted drugs and complementary medicines (CMs) for less than 2 months. The use of CMs for the management of RVVC, such as vaginal douches, probiotics (oral or vaginal), and specific herbal products such as Caprylic acid, Origanum vulgare: Oregano Hydrastis canadensis: Golden Seal, Handroanthus heptaphyllus: Pau D’arco, Allium sativum: Garlic, Pseudowintera colorata: Horopito, were grounds for exclusion except for those willing to cease and allow a 3-week washout before baseline. Participants were asked not to use antimycotics besides the approved acute RVVC treatment of 150 mg oral fluconazole when a confirmed recurrence event was identified during the study period.
Intervention
The ICTALA IMP was applied intravaginally and contained a 3% AcA 38 mg and a 2% LA 57 mg solution set into a gelatine/glycerine base (1805 mg) as a solid total 1.9 g bullet pessary with a pH between 3.85 and 3.9 in both solid and liquid form. The IMP and placebo were prepared by a compounding pharmacy in compliant laboratory conditions and were visually identical in size, colour and packaging. Labelling of the IMP only differed in batch and kit allocation numbers. The placebo control was prepared as per the IMP but without AcA and LA with pH 5.
Outcome measures
Data were collected from trial participants via fortnightly paper diary activities and telehealth with a researcher. Health assessments included licenced modified Dermatology Quality Life Index (m-DLQI) (replacing the word skin with vulva) and licenced EuroQol EQ5D-3L questionnaires, vaginal pH assessment, as well as general hygiene, sexual interaction and menstrual hygiene questions. The study assessed RVVC recurrence via participant-initiated Sobel score with symptom onset, Savvy yeast check and self-swabs for genital microscopy and culture which provided a Gram stain analysis of bacteria and identified culturable fungal microbes.
Primary feasibility outcome measures
Feasibility measures included recruitment, retention rate and compliance to study medications and assessments.
Recruitment was determined by dividing the number of eligible participants by the number of screened participants providing a ratio of screened versus enrolled participants. Retention was assessed by dividing the number of enrolled participants by the number of participants who withdrew or were lost to follow-up with descriptive analysis highlighting reasons for withdrawal.
IMP associated compliance was calculated for the total time spent in the study and as a percentage of the 6-month intervention period. Compliance at 100% constituted a total of 42 pessaries used (seven pessaries a month over 6 months). Pessary use was tracked in the diary and confirmed during the telehealth appointments with the researcher. Variances to the intervention schedule are described. Compliance to study assessments (fortnightly diary activities: hygiene questionnaire, sexual interaction questionnaire, vaginal pH recording, menstrual hygiene questionnaire, m-DLQI and EQ5D-3L) was assessed via calculation of the total data collection time points while enrolled and as a percentage of total data collection time points for the potential 9-month duration.
Secondary effectiveness outcome measures
Effectiveness measures included reduction and recurrence of RVVC episodes over the trial period, the effectiveness of IMP blinding, as well as the acceptability, satisfaction, safety and tolerability of the intervention. The feasibility of quality-of-life (QoL) measures as methods to measure effectiveness was also explored. A reduction of recurrence and severity was measured by the number of relapse events. A positive mycological episode needed to be confirmed by culture and microscopy and required treatment with a single dose of acute oral fluconazole 150 mg during the trial period. Participants needed to self-reflect on personal symptoms daily which included completing the Sobel symptom score, Savvy yeast check and self-swab for microscopy and culture when symptomatic.
Participant perceived effectiveness, satisfaction, acceptability and tolerability of the IMP were assessed at 6 months by asking participants if they thought the IMP was effective for their RVVC management if they thought they had received the active or placebo, would they recommend the IMP to others and what they liked or disliked about the IMP. After a further 3-month observation period (at the 9 months close of study phone call), participants were asked to reflect on how they currently felt about their RVVC management overall. At this time, participants were also encouraged to provide final comments on the IMP and study overall. General tolerability was additionally assessed via questioning when participants withdrew from the study and the collation of any adverse event data. The QoL measures m-DLQI and EQ5D-3L were scored fortnightly and assessed for statistical significance to determine their appropriateness as QoL assessments for this trial design. Statistical changes to these QoL measures together with changes to vaginal pH also support safety and tolerability assessment.
Randomization
A Clinical Trial Unit staff member not involved in the trial allocated and blinded participants via a block randomization schedule with computer-generated random numbers. Participants, researchers and investigators involved in the trial were blinded to the treatment allocation.
Sample size
To establish the feasibility of this study design, we aimed to recruit 30 participants to estimate the recruitment and retention outcomes. 24 Given this was a feasibility study, we did not undertake a formal sample size calculation.
Statistical methods
Descriptive statistics are presented as mean value and standard deviation (SD) for continuous variables and frequency (percentage) for categorical variables. The association between study group (active/placebo) and categorical outcomes was investigated using Fisher’s exact test. The association between study group (active/placebo) and continuous outcomes was investigated using Student’s t-test. Analyses were conducted using Stata statistical software v14 (StataCorp, College Station, TX, USA).
Results
Participant flow
Sixty-four people were prescreened via an online survey, with 43 deemed ineligible for the reasons detailed in Table 1.
Reasons for prescreening failures.

IUD: intrauterine device; IVF: in vitro fertilization.
The remaining 21 participants were further screened via phone. Three declined participation due to personal circumstances, whereas 18 completed the home screening tests. Of those, 3 were a screen failure (Savvy yeast check and Sobel symptom score indicative of active VVC infection, positive pregnancy test) and 15 (23.4%) received the study pack (Figure 2).
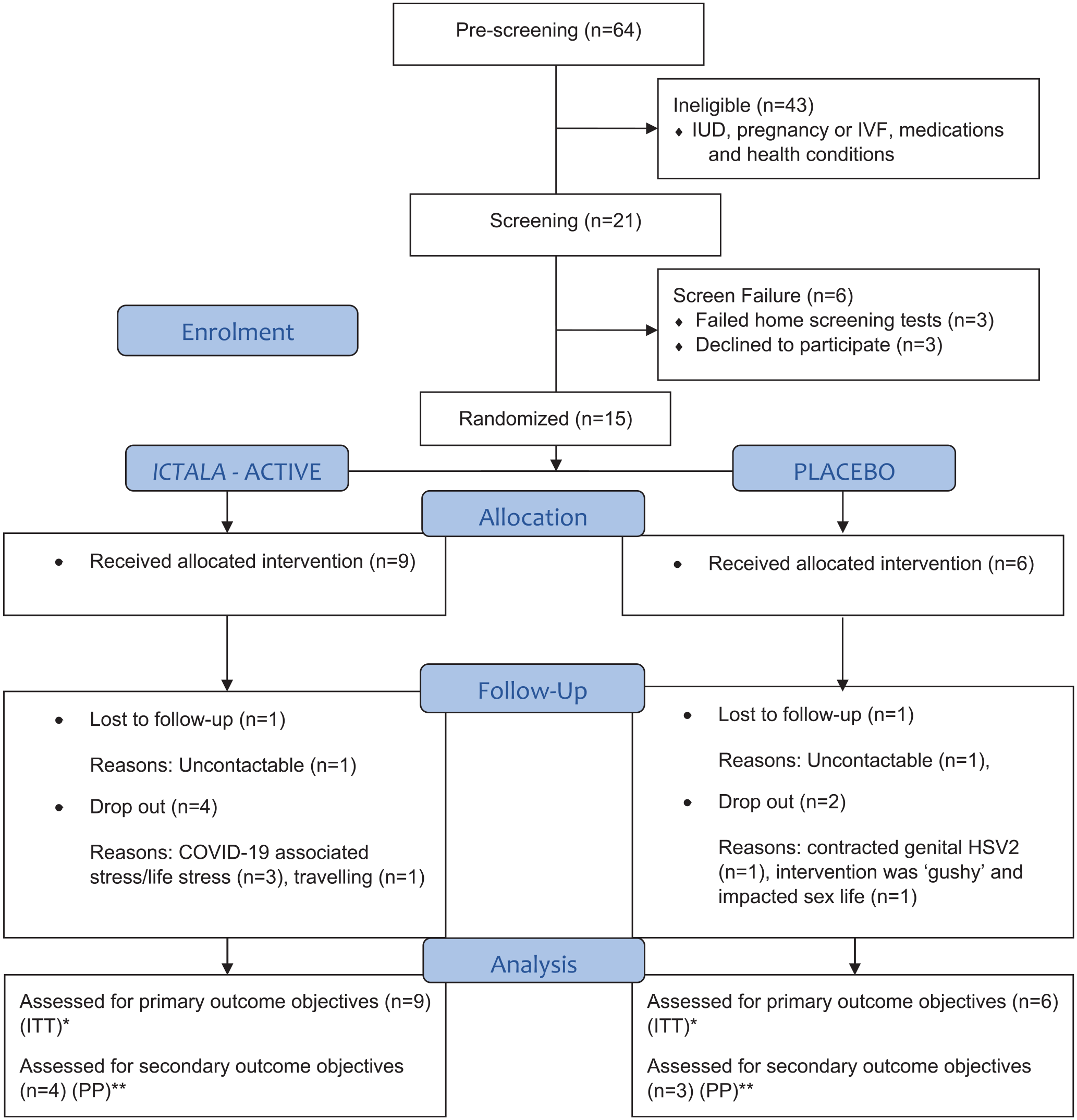
Flow chart of the trial participants (ITT and PP analyses).
Baseline characteristics
Table 2 reports on the participants characteristics. The age of the participants ranged from 20 to 47 years, with a median age of 31 years. On average, in the previous 12 months, RVVC-related symptomatic episodes occurred with mean (SD) annual incidence of 7.9 (3.1). There were no significant differences between the active and placebo groups across all variables at baseline.
Study characteristics of the total, active and placebo groups.

SD: standard deviation; RVVC: recurrent vulvovaginal candidiasis; HSV: herpes simplex virus; OCP: oral contraceptive pill.
Includes cycle equivalent for n = 3 on OCP.
Before enrolment, participants managed their RVVC with various approaches and similarly to their characteristics, there were no significant differences between participants in the active versus placebo group across all variables (Table 3). All had previously used oral fluconazole as an intermittent therapy or extended maintenance. Fluconazole was reported to resolve symptomatic episodes only to experience relapse by 93.3% of participants, with one participant reporting it had no effect (6.7%).
Past management of RVVC episodes: total, active and placebo groups.
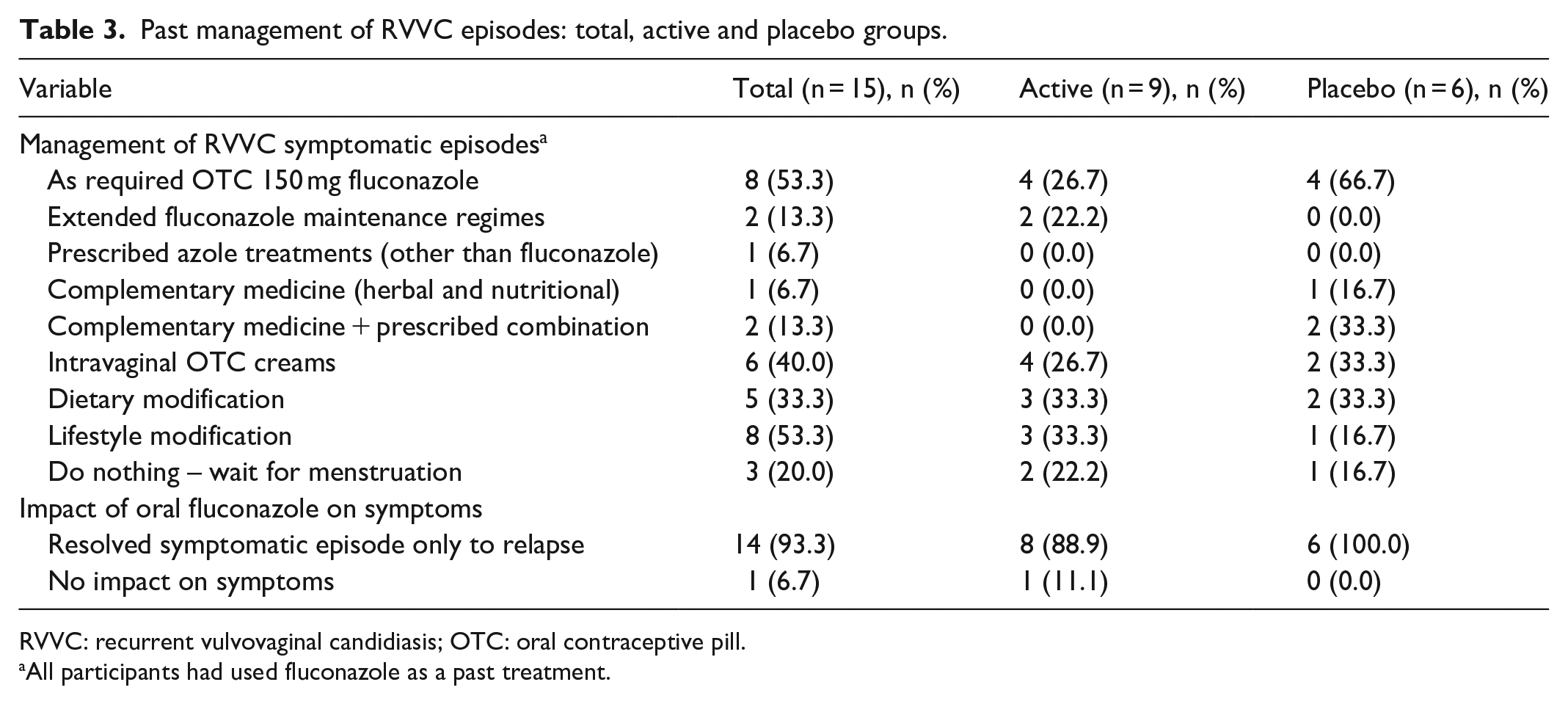
RVVC: recurrent vulvovaginal candidiasis; OTC: oral contraceptive pill.
All participants had used fluconazole as a past treatment.
Usage of and compliance to study medication
During the study, eight participants did not complete the trial and were lost to follow-up at different stages of the study (Figure 3). The remaining seven participants completed the trial and submitted data used for assessment of secondary effectiveness outcomes.

Timeline indicating withdrawals of participants.
Of those seven completing the trial, six were 100% compliant, whereas one only achieved 57% compliance. Of those eight who did not complete the trial, one withdrew during the 6-month intervention phase, although being 100% compliant until withdrawal. The other seven showed variable compliance to IMP for reasons such as (1) inconvenient to travel with IMP or to take to other places (n = 3), (2) misunderstood instructions and used fewer pessaries in the first 3 months (n = 1) and (3) busy and forgot (n = 1). For two non-completers, the reasons for non-compliance are unknown. No adverse events were recorded for any participant. Supplementary Table S1.0 illustrates the individual participant IMP compliance.
Compliance to study assessments
Eight participants did not complete the study. Two of those returned incomplete diaries for the time they participated in the study, whereas six participants did not return their diaries. Another participant completed the study submitting a diary with missing data as they had misplaced the first diary. Supplementary Table S2.0 illustrates individual participant compliance to study assessments.
Secondary effectiveness outcome measures
Reduction and recurrence of RVVC episodes over the trial period
The number and severity of RVVC symptomatic events per participant over the period of enrolment determined IMP efficacy. Four participants reported six symptomatic RVVC relapses. The mean incidence of recurrence during the study across both groups was 1 episode per year, which compared favourably with a reported mean annual incidence of 7.9 episodes reported in the 12 months prior to enrolment. There was a non-significant difference between the mean incidence rates in the active and placebo groups during the trial period (1.1 and 0.8 episodes per year, respectively). As indicated in Figure 4, two participants (active) experienced a symptomatic recurrence within the first 8 days of the trial before IMP initiation. One of these participants (1015) experienced recurrence again at day 70 (post second IMP application) associated with antibiotic use for a urinary tract infection. The same participant flared at day 90 (post third IMP application). Participant 1017, who experienced recurrence before initiating IMP use, reported a monthly frequency of severe RVVC episodes before entering the study, and participant 1015 experienced severe symptoms before the trial on average every second month. Of the two participants in the placebo group who experienced recurrence, one reported symptoms late in the treatment phase (day 159, month 6) and the other in the 3-month observational phase (day 210, months 7–9). Both participants in the placebo group who experienced recurrence reported symptom frequency of every second month before trial enrolment. All participants who experienced a recurrence during the study recorded a culture positive for C. albicans with a medium to heavy growth.
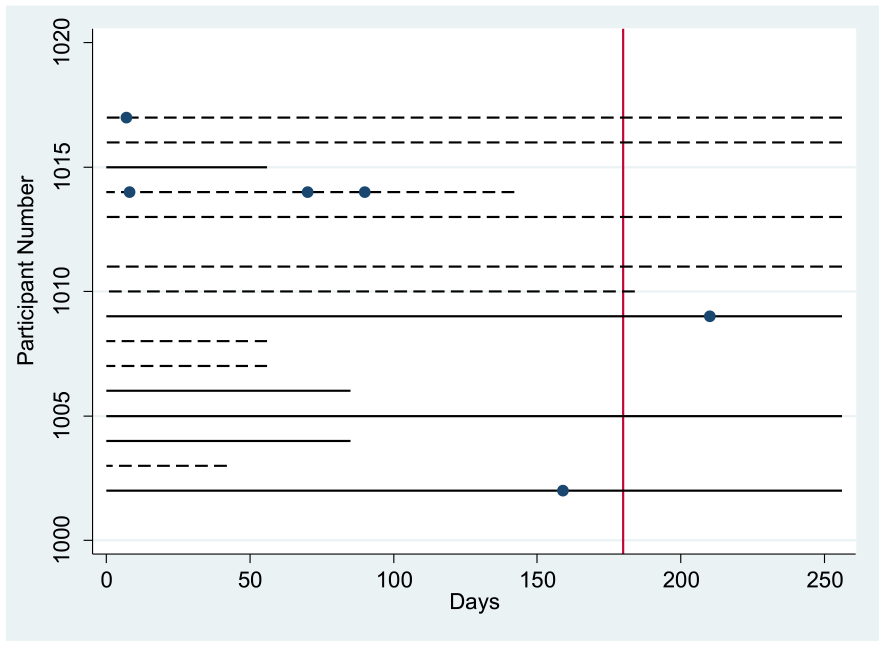
Recurrence episodes shown for individual participant. Solid lines represent the placebo and dashed lines represent the active participants. The vertical line at 170 days represents the end of intervention period. Solid circles are when recurrences occurred. The lines continue for the time the person was enrolled in the study.
Assessment of the intervention
Participant expectation of intervention effectiveness
At the 6-month phone call (day 170), all participants indicated verbally whether they believed they were using the active ICTALA or placebo IMP treatment. Sixty-three per cent of participants (n = 5 out of 8) identified their treatment allocation correctly citing symptom response as the reason for their opinion. No participants discussed the presence or absence odour as an indicator of active group involvement.
Only two participants believed they had received a placebo, with only one of them being indeed in the placebo group. Of the seven participants who withdrew or were lost to follow-up before the 6-month phone call, three were allocated to the placebo group, and four were allocated to the active ICTALA group.
Acceptability and satisfaction
All eight participants who completed the 6-month intervention phase of the study stated at the 6-month follow-up phone call that they would recommend the IMP to others. Five noted that they recommended the intervention due to a perceived decrease in symptom frequency and severity, a change to how they felt about their diagnosis, and their overall understanding of their symptoms. Three commented about the benefit of the study enabling them to try another treatment option.
Four of the seven participants who completed the full 9-month study period (intervention phase and 3-month follow-up) reported feeling better about managing their RVVC. Of these participants, all cited a reduction in symptoms as the reason for their response. Of note, only one of these participants received the placebo.
All participants in the active group who completed the study protocol expressed interest in accessing the intervention after the trial, in contrast to none in the placebo group. All participants reported the study as a positive experience:
Overall the experience was positive; the most life-changing aspect is the impact on sex, no post-sex flares. (1013) I had no symptoms through the study until I contracted COVID-19; even then it was less intense . . . and not bad enough on the Sobel score to call it a thrush flare. (1011)
One was appreciative of the body literacy the diary tracking and symptom focus provided:
The trial was a good way to understand my body better. It aided understanding of hormonal influences on my vaginal microbiome. (1002)
Feedback about the study also focussed on the intervention appropriateness:
If given the pessaries again, I would use them I felt they provided relief. In the last 3-months, feels like symptoms have come back a little. (1005)
Tolerability
All participants reported a dislike for the ‘gushy’ fluid let-down associated with the dissolution of the pessary, necessitating additional clothing or sanitary liners to minimize feeling wet. Three of the nine participants said they preferred a shorter intervention course but continued because of the intervention’s perceived benefit. One participant suggested that an applicator would be helpful for insertion.
No significant positive or negative impact on QoL using the m-DLQI and the EQ5D-3L was identified and neither was a pH change (Figure 5).

Graph of vaginal pH by days per participant. Participants allocated to the active treatment have the prefix ‘A’, while participants allocated to the placebo treatment have the prefix ‘P’.
Participant-specific vaginal pH readings reveal minor variations over the 6-month IMP phase and in the 3-month follow-up stage. Both groups recorded readings above the healthy range, for example, >4.5 at baseline and throughout the trial intervention and observation phases. Elevations of pH > 4.5 were not consistently associated with symptomatic recurrences in either group. No adverse events were reported during the study.
Feasibility of quality-of-life and economic outcome measures as methods to assess the efficacy of the intervention
There was little change in EQ5D-3L data across the study period, with most participants recording ‘1’ indicative of ‘no problem’ on most occasions. For EQ5D-mobility, all participants recorded ‘1’s, except for one participant (1009) who recorded ‘2’ (some problem) on six occasions. For EQ5D-personal care, all participants recorded ‘1’s. For EQ5D-usual activities, all participants recorded ‘1’s, except for one participant (1009) who recorded ‘2’ on six occasions. For EQ5D-pain and discomfort, six participants recorded ‘2’s, three once only, one on four occasions and two on seven occasions. Results for EQ5D-State of health, a visual analogue scale where participants rated their own health between 0 and 100 providing a numeric estimate of their health-related QoL, showed no significant between group differences at any time point in the study. Supplementary Table S3.0 provides EQ5D-State of health scores over the trial period.
m-DLQI
QoL data for the m-DLQI showed a mean initial score in the active and placebo groups of 3.0 and 4.5, respectively (mean difference = −1.5; 95% confidence interval (CI) = −7.5 to 4.5), and corresponding mean follow-up scores of 3.0 and 0.7 at the final 6-month point mean change in m-DLQI of 2.3 {% CI} which as per the tool was not statistically significant or clinically important. Supplementary Table S4.0 reports m-DLQI scores over the trial period.
Discussion
This study is the first study to examine the feasibility of a study design using an intravaginal prophylactic application of AcA and LA to prevent RVVC symptomatic episodes in an at-risk population. The study results provide insight into design feasibility and identify significant recruitment challenges and obstacles for high attrition. Design and implementation adaptation in response to challenges encountered during recruitment resulted in enhanced recruitment in the later stages of the recruitment period. The attrition rate of 40% at 6 months and 53% at 9 months is similar to rates of other trials using intravaginal applications in RVVC. 25 Overall, participants exhibited high compliance to the treatment and trial procedures indicating that the intervention use and frequency of application were feasible and acceptable to participants.
Recruitment challenges
The initial trial design captured people as they transitioned off fluconazole maintenance therapy to improve the post-therapy relapse rate; however, early recruitment efforts and qualitative interviews of Australian RVVC patients revealed that access to and prescription of maintenance therapy is nuanced and inconsistent 1 and that this eligibility criterion limited recruitment unnecessarily. A decision was made to recruit a broader RVVC population independently of their fluconazole use. On further review, other eligibility criteria were adapted including participants with medication controlled Hashimotos or a latent genital herpes simplex virus (HSV1 and HSV2) diagnosis to enhance recruitment.
Twelve per cent of the screened population were ineligible because they did not have a formal diagnosis, including being able to demonstrate adequate culture-positive symptomatic episodes. This aligns with a qualitative study that showed obtaining a definitive RVVC diagnosis is problematic within the Australian health setting. 1 During the screening, 26% of people were found to be ineligible for having an IUD such as the Mirena, copper coil or for using a POC. The potential interaction of the acidic IMP, on IUD structure and function as well as the possible change to recurrence from POCs and IUDs underpinned this eligibility criterion. Theoretically, LA and AcA would be inert when in contact with IUDs as the ICTALA IMP pH was in alignment with that of a healthy vaginal pH (3.8–4.5); however, no definitive safety studies exist exploring IUD composition and material stability. This research gap should be evaluated before including people with IUDs into future trials evaluating ICTALA.
IUDs are associated with an increased risk of BV and VVC,26,27 whereas POCs are believed to reduce RVVC risk. 28 Given the potential prescription of POCs in RVVC for symptom relief, inclusion of people taking POCs and an expansion of the eligibility criteria to allow those with excluded health conditions, including a history of mixed infections (BV and VVC), would allow a greater generalizability of the results to the RVVC population.
Overall, the absence of a formal feasibility sample size calculation, recruitment challenges and resulting lower than expected participant numbers limit the interpretation and generalizability of the secondary outcomes related to ICTALA IMP efficacy and changes to participants’ QoL assessments. The projected sample size to achieve power in a full clinical trial would require considering obstacles to recruitment and modifying eligibility criteria.
Retention and COVID-19 impacts
COVID-19 presented challenges in the execution of the study, initially by impacting recruitment and face-to-face participant interaction. Challenges were responded to by adapting recruitment criteria and trial design to a telehealth model encompassing a national participant pool. Participants who enrolled at the height of the COVID-19 crisis and amid Australian lockdowns had a higher dropout rate than those who enrolled later in the pandemic, for example, in 2021 and early 2022. The actual reasons for this are difficult to determine. However, two participants noted that life stress was a factor in their withdrawal, with one citing extreme COVID-19-associated stress with Victorian (Australia) extended lockdowns. Overall, while recruitment limitations were not all COVID-19 associated, the overall influence on trial exposure, healthcare practitioner access and participant willingness to engage due to the global COVID-19 crisis may not be as impactful in future trial planning and feasibility assessment.29 –31
Adherence to trial procedures and data collection limitations
Participants who remained enrolled for trial duration had a high adherence to trial procedures. The trial used paper diaries for data collection and trial progression tracking was initially chosen due to the planned face-to-face nature of the study. Questioning at the 6- and 9-month calls revealed that participants would prefer digital diaries. Data collection through online interfaces and specifically designed mobile applications is superior to paper collection, providing a higher proportion of diaries with completed to-date (of enrolment duration) data and contextual information.32,33
Recurrence assessment
This feasibility study was not powered to detect a statistical reduction in symptomatic occurrences associated with the IMP. When viewed individually and as episodes over time, all participants who completed the study experienced a change in the number of symptomatic episodes. Participants in the ICTALA group showed sustained treatment effects, with continuing absence of symptoms during the IMP phase and through the 3-month post-IMP follow-up (9 months), except for one participant who experienced recurrence with antibiotic use and later withdrew. Lack of symptomatic episodes 3-month post-IMP contrasts to a high relapse rate of symptomatic RVVC shortly after the cessation of weekly maintenance therapy with fluconazole. 21 Participants in the placebo group also noted a reduction in symptom frequency while using the IMP. It is difficult to determine whether the change to recurrence was associated with the intervention itself, a possible placebo effect 34 or related to other non-IMP factors such as psychological support provided through appropriate disease-specific communication and clinical attention during the trial.
Qualitative discussions with participants revealed that receiving support during the trial through disease-specific communication, education and empathy may have considerably impacted their symptom awareness and how participants perceive their condition. This aligns with research that suggests positive and informed healthcare experiences could improve patient outcomes. 35 In addition, increased focus on menstrual cycle timing, symptom risk periods and subsequent potential changes to sexual interaction timing or hygiene have been shown to decrease recurrence. 27
Currently, there is not a specific questionnaire that accurately assesses the consequences of RVVC on the QoL and sex life of people with RVVC; however, one is reported to be in development. 2 Future trial design incorporating a measurable framework of benefits of participant support, with a focus on psychological health, is warranted. This aligns with recommendations that RVVC patients receive psychological referrals as part of their long-term management. 1 Psychological support could be assessed through a comparative arm where participants receive specific and holistic one-on-one psychological intervention rather than IMP. In addition, for a more conclusive analysis of changes to recurrence, a comparative arm that employs a commonly used treatment, such as vaginal clotrimazole or oral fluconazole, could be utilized.
Suitability of quality-of-life measures
Prior studies assessing the health-related QoL of individuals with RVVC have demonstrated that subjective health status and QoL are diminished not only during acute symptom episodes but also outside of symptomatic episodes.8,36,37 In this study, the use of QoL measures m-DLQI and EQ5D-3L failed to demonstrate a significant change in QoL domains during the trial across both groups. These outcome measures showed minimal variability, and statistically, participants exhibited hallmarks and scores across assessed domains in both the m-DLQI and the EQ5D-3L with a high QoL. The trial recruited people with RVVC who were asymptomatic on enrolment, as preventing RVVC symptomatic relapse was one of the measurable trial outcomes. In other RVVC trials where QoL outcome measures have demonstrated statistically significant change, participants were usually symptomatic when recruited, assessed, treated and assessed again when symptoms resolved.38 –40 Thus, scores achieved in this study should be interpreted as maintenance of wellness rather than a measurement of change in disease severity. When viewed this way, minimal variance to individual domain scores is significant with the overall scoring and reported state of health, consistent with good QoL for participants.40,41 Future large-sized trial design should consider and interpret the maintenance of the QoL scores and/or look for more sensitive measures of wellness maintenance specific to RVVC. Prior qualitative research demonstrated that the RVVC impacts on QoL were physical and psychological, 1 often relating to frustration associated with poor disease management, unsympathetic healthcare practitioners and the realization of having a lifelong condition. 1 This study not only provided the hope of a potential new treatment option but also provided education and information about RVVC from sympathetic and informed researchers, the impacts of which cannot be overlooked for its potential to have a beneficial psychological effect and as such to decrease the RVVC disease burden.
Strengths and limitations
The study’s strengths included factors often overlooked in RVVC trial design as identified by a recent Cochrane review on the treatment for RVVC, including the collection of outcomes relevant to patients such as the mean number of recurrences in a time period, symptom-free days before recurrence and information around patient treatment preferences. 4 The study also collected data about recurrence without the restrictions of defined time points or scheduled visits for swabbing, 4 for example, participants were encouraged to self-reflect daily on symptoms and if feeling symptomatic complete the recurrence confirmation steps which included self-swabbing to confirm mycological recurrence. This allowed an accurate capture of recurrence specific to the individual. The timing of the IMP to introduce a preventive and vaginal environment influencing treatment prior to a high-risk RVVC relapse time (mid luteal) 42 was also seen as a strength of the study design. The majority of RVVC studies that explore maintenance and intravaginal prevention interventions either apply treatment after menstruation or on a weekly frequency. 43 This leads to potentially missing the opportunity to modify the environment and mycotic biorhythm directly prior to the highest risk period. 44 Future adaptations of the study design could utilize variations of the ICTALA IMP dosage form to assess patient preferences.
This study is a feasibility trial and displayed the limitations and problems exhibited by such investigations with low sample size and subsequent impacts on statistical significance. In addition, low recognition and diagnosis of RVVC in clinical practice previously reported 1 could have played a role in the recruitment difficulties. This was amplified with the unexpected pressures of the COVID-19 global pandemic.
Conclusion
The protocol was feasible to assess the recurrence impacts of an intravaginally applied prophylaxis therapy. In a future larger-sized trial, streamlined digital data collection would support a more effective evaluation of intervention-associated efficacy outcome measures. This would allow the collection of completed to-date data from participants lost to follow-up or early withdrawal. Recruitment numbers could be improved in a post-acute COVID-19 landscape. Expansion of recruitment eligibility to include those with IUDs, various oral contraceptive types and mixed infections and engaging multi-medical practice locations may serve a multi-purpose by recruiting a broader RVVC community, allowing broader transferability of data in the RVVC population and increasing the recognition of RVVC as a diagnosis.
Re-considering which QoL measures are used would be appropriate; with a larger and broader sample size, the chosen measures m-DLQI and EQ5D-3L may provide meaningful results; however, the utilization of a wellness maintenance measure may be more appropriate. Based on positive participant feedback and the observed reduction in episode recurrence, the efficacy of ICTALA should be explored. This includes a need for more insight into the impact of psychological and clinician support in RVVC on symptom severity and recurrence. A larger full-scale trial is warranted.
Supplemental Material
sj-docx-1-whe-10.1177_17455057231194138 – Supplemental material for Intravaginal Combination Therapy of Acetic and Lactic Acid in premenopausal women with recurrent vulvovaginal candidiasis: A randomized, double-blind placebo-controlled feasibility trial
Supplemental material, sj-docx-1-whe-10.1177_17455057231194138 for Intravaginal Combination Therapy of Acetic and Lactic Acid in premenopausal women with recurrent vulvovaginal candidiasis: A randomized, double-blind placebo-controlled feasibility trial by Moira Bradfield Strydom, Sohil Khan, Ramesh L Walpola, Chris Testa, Robert S Ware and Evelin Tiralongo in Women’s Health
Supplemental Material
sj-docx-2-whe-10.1177_17455057231194138 – Supplemental material for Intravaginal Combination Therapy of Acetic and Lactic Acid in premenopausal women with recurrent vulvovaginal candidiasis: A randomized, double-blind placebo-controlled feasibility trial
Supplemental material, sj-docx-2-whe-10.1177_17455057231194138 for Intravaginal Combination Therapy of Acetic and Lactic Acid in premenopausal women with recurrent vulvovaginal candidiasis: A randomized, double-blind placebo-controlled feasibility trial by Moira Bradfield Strydom, Sohil Khan, Ramesh L Walpola, Chris Testa, Robert S Ware and Evelin Tiralongo in Women’s Health
Footnotes
Acknowledgements
The authors thank and acknowledge the participants for their time and willingness to share data about their daily lives. The authors thank Tugun Compounding Pharmacy for their support in co-development of the ICTALA formulation and IMP production, Ariya Health for their supply of pH strips and Elisabeth Kolarich for her support with IMP liaison, blinding and randomization.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.